
Tras muchos años de ensayos clínicos fallidos, un estudio llevado a cabo en 12 pacientes, usando una novedosa terapia génica desarrollada por la empresa biotecnológica BioMarin Pharmaceuticals, ha mostrado esperanzadores resultados en el tratamiento resolutivo de la hemofilia tipo A (la que cursa con déficit del factor IX de la coagulación). Solo dos de los doce pacientes sujetos de estudio no respondieron al tratamiento.
Esta terapia consiste en insertar un gen en el genoma de los pacientes hemofílicos mediante un virus. Los tratamientos génicos de la hemofilia, tipos A o B, son terapias a «todo o nada». Si no funcionan, el paciente se ve obligado a reiniciar las trasfusiones periódicas de los factores de coagulación de los que carece [La hemofilia tipo B se conoce también como enfermedad de Christmas. Existe una variante, hemofilia tipo B Leyden, que afecta a los niños y suele mejorar o solucionarse a partir de la pubertad].
Genética de la hemofilia
 La prevalencia de la hemofilia tipo A se estima en 1 caso cada 4.000 o 5.000 varones nacidos vivos; mientras la prevalencia de hemofilia tipo B es de 1 caso por cada 20.000 varones nonatos.
La prevalencia de la hemofilia tipo A se estima en 1 caso cada 4.000 o 5.000 varones nacidos vivos; mientras la prevalencia de hemofilia tipo B es de 1 caso por cada 20.000 varones nonatos.
El rasgo hemofílico se transmite como herencia recesiva ligada al sexo (cromosoma X). La enfermedad la padecen los varones; las hembras actúan como portadoras asintomáticas.
Así, la hemofilia de un abuelo (varón) que no se manifiesta en ninguno de sus hijos varones, tiene una probabilidad del 50% de aparecer en sus nietos varones.
La hija de una portadora tiene una probabilidad del 50% de transmitir la enfermedad a sus hijos varones.
Características de la herencia recesiva ligada al sexo
- La incidencia del rasgo es mucho mayor en ♂ que en ♀.
- Las ♀ heterocigóticas no están afectadas; o lo están parcialmente en función del patrón de inactivación del cromosoma X.
- El gen responsable se transmite de un ♂ afectado a través de todas sus hijas (♀). Los descendientes varones de sus hijas tienen una probabilidad del 50% de heredar la condición.
- El gen nunca se transmite de padre a hijo varón (♂).
- El gen se transmite de varón a todas sus hijas (♀).
- El gen se perpetúa en sucesivas generaciones a través de las mujeres.
- Aunque infrecuentes, existen mutaciones «de novo».
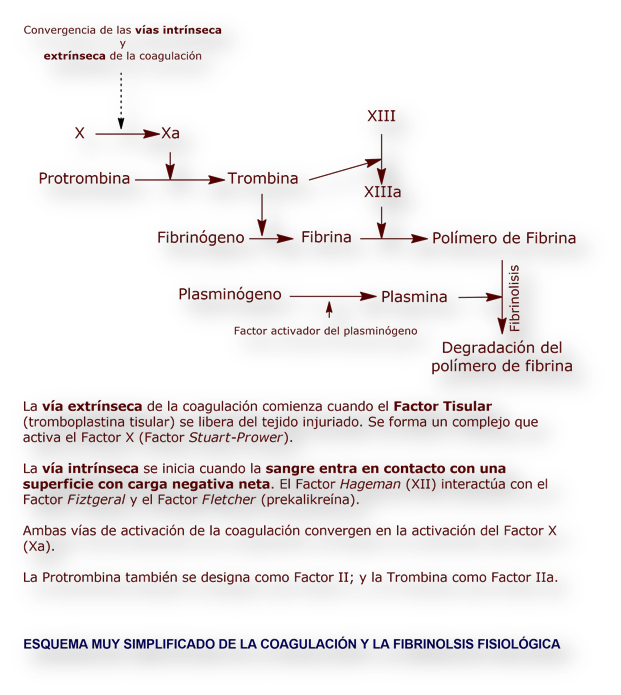
La hemofilia reveló las primeras etapas de la que más tarde se pergeñó como «cascada de coagulación»: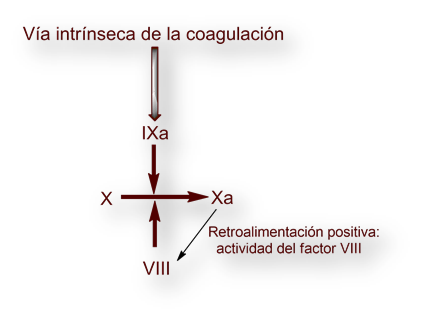
En la «hemofilia clásica» o hemofilia tipo A, el factor VIII (el único de la «cascada de coagulación» que no tiene actividad enzimática tipo proteasa) está ausente o tiene una actividad residual. El factor VIII actúa como adyuvante necesario para la activación del factor X por el factor IXa (factor IX activado). [La ausencia del factor IX es la causa de la hemofilia tipo B o enfermedad de Christmas].
Los hemofílicos más gravemente afectados necesitan la administración intravenosa cada pocos días del factor de coagulación que no pueden sintetizar, en función de si están afectados por hemofilia tipo A o B. Estas proteínas tienen Vidas Medias muy breves, haciendo necesarias inyecciones periódicas. A pesar del tratamiento, los hemofílicos continúan teniendo riesgo de sufrir hemorragias «espontáneas», tras mini-traumatismos imperceptibles incluso para el paciente.
 Si bien el cerebro es el órgano más comprometido, las hemorragias pueden dañar irreversiblemente otras estructuras corporales, tales como las articulaciones. Es común que los hemofílicos se hayan de someter en edades tempranas a cirugía protésica de rodilla o cadera. El dolor crónico es común en estas personas.
Si bien el cerebro es el órgano más comprometido, las hemorragias pueden dañar irreversiblemente otras estructuras corporales, tales como las articulaciones. Es común que los hemofílicos se hayan de someter en edades tempranas a cirugía protésica de rodilla o cadera. El dolor crónico es común en estas personas.
Los hemofílicos confrontan múltiples problemas durante su vida diaria. Cuando niños, han de evitar deportes de contacto físico, en prevención de hemorragias internas o externas. Más adelante, sus opciones laborales se hallan así mismo restringidas. Por otra parte, sus seguros médicos privados conllevan unos costes prohibitivos.
Durante la década de 1980, los factores de coagulación que recibían los hemofílicos estaban contaminados por Virus de Inmunodeficiencia Humana (VIH), y virus de la hepatitis C. La mayoría de las personas hemofílicas de entonces se infectaron, y un sinnúmero fallecieron por estas causas.
Por suerte, podemos hallarnos ante un avance trascendente en el tratamiento resolutivo de la hemofilia: la terapia génica.
Conceptualmente, la terapia génica de la hemofilia consiste en insertar el gen deficitario o defectuoso. Si el tratamiento tiene éxito, se evitan las hemorragias frecuentes, y la necesidad de continuas inyecciones de factores de coagulación, hoy obtenidos por ingeniería genética. Aun cuando los síntomas son similares, la hemofilia tipo A (hemofilia clásica) está causada por ausencia o grave deficiencia de actividad factor VIII, mientras la hemofilia tipo B (enfermedad de Christmas) se debe a la ausencia del factor IX.
Dos empresas farmacéuticas han iniciado ensayos clínicos con sus terapias génicas: Spark Therapeutics junto con Pfizer, estudia su terapia contra la hemofilia tipo B, usando SPK-9001 (Fidanacogene elaparvovec); y BioMarin Pharmaceuticals ensaya AAV5-hFVIII-SQ . (Valoctocogene roxaparvovec) contra la hemofilia tipo A.
Los comienzos de la terapia génica de la hemofilia estuvieron trabados de dificultades.
Los pacientes con hemofilia tipo A tratados con AAV5-hFVIII-SQ (terapia génica desarrollada por BioMarin Pharmaceuticals) consiguieron el primer año niveles normales de factor VIII en suero, que se redujeron una mediana del 46% durante el segundo año.
En cambio, los pacientes con hemofilia tipo B tratados con SPK-9001 (la terapia génica de Spark Therapeutics) lograron un promedio del 35% de los niveles considerados normales de factor IX. A pesar del más modesto éxito inicial, éste se mantuvo a lo largo del tiempo de seguimiento (dos años). Este 35% de la actividad considerada normal del factor IX parece ser suficiente para la prevención de hemorragias en las personas con hemofilia tipo B.
La hemofilia parecía una genopatía adecuada para el abordaje terapéutico mediante terapia génica. De una parte, los niveles séricos normales de las proteínas (factores) de coagulación tienen una amplia mediana, del 50% al 150%. De este modo, incluso una terapia génica con modestos resultados podría ser efectiva. Además, los genes involucrados en los dos tipos de hemofilia (A y B) se conocen desde los primeros años de la década de 1980.
Los primeros resultados con la terapia génica en el tratamiento correctivo de la hemofilia se obtuvieron hace aproximadamente una década en la University College of London. Un experimento llevado a cabo en diez pacientes con hemofilia tipo B, consiguió un incremento de los niveles de factor IX de entre un 2 y un 6%. Aunque el incremento fue modesto, estos niveles se mantuvieron relativamente constantes desde entonces.
Inesperadamente, se halló a un hombre de Padua, Italia, portador de una mutación genética que hace que sus células sinteticen hasta 12 veces las cantidades usuales de factor IX (factor Christmas).
La excepcional mutación del gen, una vez clonado, se insertó en el genoma de un virus, que actuó como transportador del gen a las células de los pacientes con hemofilia tipo B.
La ventaja es que un gen más potente permitía usar una menor «carga viral» y, de esa manera, restringir la posible respuesta del sistema inmune del paciente.
La primera paciente así tratada fue una enfermera de 23 años. Sus niveles de factor IX se incrementaron alrededor de un 30%, permaneciendo constantes desde entonces. La paciente no necesitó más transfusiones de factor IX, ni sufrió hemorragias espontáneas.
Sin embargo, el desarrollo de la terapia génica de la hemofilia tipo A ha sido mucho más difícil.
Los virus utilizados para introducir genes en las células de los pacientes son adenovirus. Éstos no pueden transportar genes de gran tamaño; y el gen que codifica el factor VIII lo es. [El gen del factor VIII abarca 186 Kb, distribuidos en 26 exones (que ocupan 70,5Kb), por lo que la mayor parte del gen está formado por intrones. La proteína expresada por este gen está formada por 2.351 aminoácidos con un peso molecular total de 265.000 daltons, antes de su procesamiento].
Durante tres lustros, la investigación se ha dirigido a eliminar aquellas partes innecesarias del gen para poder insertarlo en un adenovirus transportador.
Nos hallamos ante la posibilidad de curación de una genopatía que ha determinado, como ninguna otra, el devenir de la Historia. Durante muchos años se le denominó «hemofilia real», al afectar a muchos herederos de monarquías europeas.
Zaragoza, a 20 de agosto de 2018
Dr. José Manuel López Tricas
Farmacéutico especialista Farmacia Hospitalaria
Farmacia Las Fuentes
Florentino Ballesteros, 11-13
50002 Zaragoza