
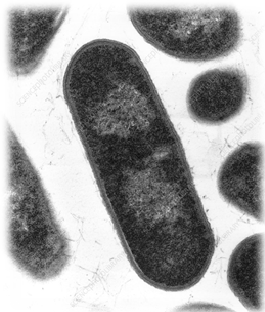
Células de un cáncer pancreático (ampliación bajo microscopía electrónica)
Un tratamiento personalizado contra el cáncer de páncreas avanzado y metastásico, llevado a cabo en dos mujeres ha tenido resultados opuestos: mientras en una de ellas el cáncer desapareció, la otra no respondió y terminó falleciendo.
En ambos casos, se habían probado todos los tratamientos habituales para este tipo de carcinoma (cirugía usando la técnica de Whipple, radioterapia y quimioterapia). El experimento se ha publicado en la revista The New England Journal of Medicine[i]. La técnica usada podría ser extrapolable a otros cánceres, tales como los de pulmón, y colon y recto.
El tratamiento consistió en la reprogramación genética de las células T del paciente que, este modo, reconocen y destruyen las células cancerosas. La técnica ha sido diseñada por Eric Tran y Rom Leidner del Instituto de investigación Earle A. Chiles, adscrito al Providence Cancer Institute, en Portland, Oregón, Estados Unidos.
Conceptualmente se trata de convertir las células T del paciente en fármacos vivos.
El cáncer de páncreas suele ser muy refractario a los tratamientos convencionales; incluso con los tratamientos habituales, la supervivencia a 5 años apenas supera el 10%. Ello se debe a que, cuando se manifiesta la sintomatología, el cáncer [de páncreas], de sólito, ya ha dado lugar a metástasis. Aun cuando se produzca la exéresis del tumor primario, alrededor del 85% de los pacientes sufren recidivas.
La tecnología usada en estas dos pacientes no está disponible, por ahora, para su empleo generalizado.
El primer problema es que las proteínas mutadas que impulsan el crecimiento del cáncer se ocultan dentro de las células malignas. El «talón de Aquiles» de las células cancerosas está formado por fragmentos de estas proteínas mutadas cuando se exponen sobre la superficie celular.
La técnica consiste en extraer células T del paciente y reprogramarlas genéticamente para que reconozcan como antígenos a los fragmentos de estas proteínas mutadas cuando se expresan en la membrana de las células cancerosas. Acto continuo, cultivar las células T genéticamente modificadas, inyectándolas, vía intravenosa, en el paciente.
En este caso la proteína mutada se designa como KRAS que se sintetiza en aproximadamente uno de cada cuatro pacientes con cualquier tipo de cáncer; pero en el 95% aproximadamente de todos los cánceres de páncreas, el 40% de todos los carcinomas de colon y recto; y el alrededor de la tercera parte de todos los cánceres de pulmón. [KRAS es el acrónimo en inglés de Kirsten RAt Sarcoma Virus, codificada por un gen 12p12.1. La proteína KRAS es una enzima que cataliza la conversión del GTP ® GDP]. [GTP es el nucleótido Guanina Trifostato; GDP es el nucleósido Guanina Difosfato].
Durante las últimas dos décadas se ha tratado de atacar inmunológicamente a la proteína KRAS en la esperanza de que constituyese el «talón de Aquiles» de las células cancerosas que expresan dicha proteína mutada.
La respuesta de las metástasis depende de donde se produzcan: las que aparecen en el pulmón son más susceptibles que las hepáticas.
Una cuestión trascendente es por qué de las dos pacientes con cáncer de páncreas con metástasis generalizadas, que se habían mostrado refractarias a los tratamientos habituales, una respondió de modo espectacular, mientras en la otra la enfermedad progresó y la paciente terminó falleciendo.
En los últimos años se han desarrollado progresos en la investigación del cáncer. He aquí algunos:
- La diabetes de aparición tardía (diabetes tipo 2) puede ser una señal de advertencia temprana de cáncer de páncreas incipiente.
- La quimioterapia está perdiendo predicamento a favor de la inmunoterapia, si no en todos, sí en muchos tipos de cáncer, sobre todo en los de mama y pulmón.
- Un tratamiento experimental contra el cáncer de próstata consiste en la inyección de moléculas radiactivas que localizan y destruyen las células del tejido tumoral.
- El tratamiento CAR-T para pacientes con leucemia mielógena crónica ha mostrado resultados espectaculares en dos pacientes. Este tratamiento crea esperanzas a la vez que genera nuevas interrogantes.
- Nivolumab, un anticuerpo monoclonal, ha mostrado prolongar la supervivencia en pacientes con cáncer de esófago, el séptimo más prevalente en todo el mundo.
Zaragoza, a 29 de junio de 2022
Dr. José Manuel López Tricas
Farmacéutico especialista Farmacia Hospitalaria
Farmacia Las Fuentes
Zaragoza
[i] N Engl J Med 2022; 386: 2112-2119












{kind=link}