
En el año 1907, Windaus y Vogt sintetizaron la histamina en el laboratorio. Tres años después, Ackermann aisló la histamina tras la fermentación bacteriana del aminoácido histidina.
 En esa misma época, Henry Dale, farmacólogo londinense, estudiaba extractos del ergot (el esclerocio del hongo Claviceps purpurea que infesta las espigas del centeno) por su posible aplicación en el control de las hemorragias postparto. Supuso, acertadamente, que una sustancia, no aislada hasta entonces, sería responsable de gran parte de la contracción de la musculatura uterina. Fue así como demostró que el «ergotinum dialysatum» de Wernich desencadenaba la contracción del útero de modo mucho más eficiente que el propio ergot. Junto al químico, George Barger, aislaron una pequeña molécula con carácter básico que reproducía por sí sola las acciones del «ergotinum dialysatum» en experimentos llevados a cabo en útero de rata. En el año 1910, Dale y Laidlaw desentrañaron la estructura de esta molécula, un β-imidazol etilamina, denominado desde entonces histamina.
En esa misma época, Henry Dale, farmacólogo londinense, estudiaba extractos del ergot (el esclerocio del hongo Claviceps purpurea que infesta las espigas del centeno) por su posible aplicación en el control de las hemorragias postparto. Supuso, acertadamente, que una sustancia, no aislada hasta entonces, sería responsable de gran parte de la contracción de la musculatura uterina. Fue así como demostró que el «ergotinum dialysatum» de Wernich desencadenaba la contracción del útero de modo mucho más eficiente que el propio ergot. Junto al químico, George Barger, aislaron una pequeña molécula con carácter básico que reproducía por sí sola las acciones del «ergotinum dialysatum» en experimentos llevados a cabo en útero de rata. En el año 1910, Dale y Laidlaw desentrañaron la estructura de esta molécula, un β-imidazol etilamina, denominado desde entonces histamina.
La histamina da lugar a vasodilatación, contracción del músculo liso (de los bronquios, intestino y útero), tiene efecto cronotropo e inotropo; y desencadena una reacción de tipo «anafilactoide» cuando se inyecta en animales.
Durante los años siguientes, la nueva molécula se estudió con profusión.
En el año 1920 Popielski dio cuenta que la histamina estimula la secreción ácida hidroalcohólica en estómago de perros.
Lewis y Grant describieron en 1924 la denominada «triple respuesta» tras la inyección subcutánea de la histamina: enrojecimiento (debido a vasodilatación), induración (por aumento de la permeabilidad a las células inmunitarias) y reflejo axónico.
Sin embargo, continuaba sin estar claro si la histamina era un mediador químico con efectos sistémicos. El grupo de trabajo de C. H. Best, en 1927, aisló histamina pura (en forma cristalizada) a partir de los tejidos hepático y pulmonar, evidenciando por primera vez que la histamina está fisiológicamente presente en todo el organismo.
Unos años más tarde se demostró su trascendente función en las reacciones anafilácticas.
En 1952, Riley y G.B. West demostraron que los mastocitos eran las principales células productoras de histamina; siendo los basófilos (una estirpe de leucocitos) la principal fuente de histamina de las células sanguíneas.
Receptor H1 de la histamina. Surgimiento de los antihistamínicos H1.
Daniel Bovet inició en 1937, mientras trabajaba en el laboratorio Ernest Fourneau adscrito al Instituto Pasteur de París, una investigación dirigida a descubrir moléculas que pudiesen bloquear alguna, o varias, de las acciones fisiológicas de la histamina. E. Fourneau sintetizaba moléculas candidatas, y D. Bovet realizaba los ensayos farmacodinámicos. Uno de los primeros hallazgos fueron dos moléculas (compuestos 883F y 933F), con estructura de benzodioxano. Se mostraron efectivas para prevenir el shock anafiláctico en cobayas. Siguiendo esta línea de investigación se sintetizaron los compuestos 929F (timoxietildietilamina) y 1571 F (N, N’-dietil-N’-fenil-N’-etiletilendiamina). Aun cuando ambos compuestos tenían una toxicidad inaceptable para su empleo en terapéutica, sirvieron de punto de partida para futuras moléculas.

Rhône-Poulenc comercializó en el año 1942 Antergan® (Fenbenzamina), modificado más tarde por su derivado Pirilamonio maleato, comercializado como Neo Antergan®, durante algunos años el único medicamento antihistamínico (H1) disponible.
Receptor H2 de la histamina. Primer antagonista H2.
Durante la década de 1940 se observó que algunos efectos de la histamina eran refractarios a los antihistamínicos H1, en particular las accione sobre el corazón (cronotropo e inotropo) y la estimulación secretora ácida del estómago. Se infirió la existencia de un segundo receptor para la histamina. La primera observación surgió tras el hallazgo del grupo de trabajo de Folkow (1948) de que Benadryl® (un clásico antihistamínico H1) podía bloquear el efecto vasodilatador de bajas concentraciones de histamina, pero no cuando las concentraciones de histamina eran elevadas. [Hay que buscar la explicación en la existencia de más de un receptor con afinidades distintas]. Un año antes (1947), Hans Schild había desarrollado un modelo matemático, designado como «valor pA2», para evaluar la acción antagonista de las moléculas, particularmente fármacos. Mediante esta metodología (pA2), Schild y Ash mostraron que el receptor H1 antagonizado por Meperidina y Difenhidramina intermedian los efectos de la histamina sobre el tono muscular intestinal y uterino.
 El descubrimiento del segundo receptor de la histamina (H2) está inextricablemente unido a James Black, Premio Nobel de Fisiología y Medicina en 1988, ex aequo Gertrude B. Elion y George H. Hitchings. J. Black dedujo que los potenciales antagonistas del receptor H2 de la histamina, tendrían una estructura química más parecida a la molécula de histamina que los antihistamínicos H1 (químicamente muy disimilares de la molécula de histamina). Partiendo de esta suposición se sintetizó la primera molécula que mostró acciones antagonistas sobre el receptor H2. Se trataba de la Burimamida. El descubrimiento se publicó en la revista Nature[1] en 1972.
El descubrimiento del segundo receptor de la histamina (H2) está inextricablemente unido a James Black, Premio Nobel de Fisiología y Medicina en 1988, ex aequo Gertrude B. Elion y George H. Hitchings. J. Black dedujo que los potenciales antagonistas del receptor H2 de la histamina, tendrían una estructura química más parecida a la molécula de histamina que los antihistamínicos H1 (químicamente muy disimilares de la molécula de histamina). Partiendo de esta suposición se sintetizó la primera molécula que mostró acciones antagonistas sobre el receptor H2. Se trataba de la Burimamida. El descubrimiento se publicó en la revista Nature[1] en 1972.
Burimamida sirvió de punto de partida para la síntesis de trascendentes medicamentos que cambiaron para siempre el tratamiento (hasta entonces exclusivamente quirúrgico) de la úlcera péptica.
El primer medicamento antihistamínico H2 fue la Cimetidina[2] (Tagamet®) en 1977. Con un período de cinco años se comercializaron Ranitidina[3] (Zantac®) en 1982; y Famotidina (Tamín®) en 1987. Los medicamentos antihistamínicos H2 para el tratamiento de las úlceras gastroduodenales, en su momento verdaderamente revolucionarios, han quedado casi relegados con el advenimiento de los «inhibidores de la bomba de protones» (véase informe sobre este grupo de fármacos en la página web www.info-farmacia.com).
HISTAMINA EN EL TEJIDO CEREBRAL
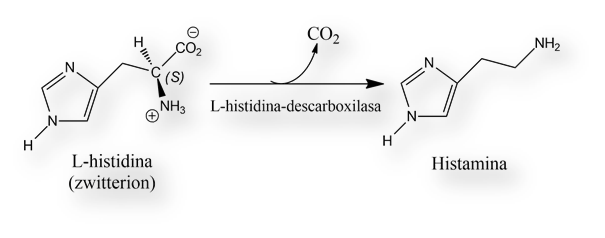
Cuando se introdujo Antergan® (Fenbenzamina) enseguida se hizo evidente que daba lugar a sedación; y que se trataba de un «efecto de grupo», extrapolable al resto de los antihistamínicos H1. Partiendo de este hallazgo clínico, Kwiatkowski en 1942 descubrió la presencia fisiológica de histamina en el tejido cerebral. Con el subsiguiente desarrollo de anticuerpos contra la enzima que cataliza la descarboxilación (L-histidina ® histamina), se rastrearon las regiones cerebrales con elevada densidad de neuronas histaminérgicas y sus proyecciones (axones) a otras áreas. La mayor densidad de neuronas histaminérgicas se localiza en el núcleo tuberomamilar del hipotálamo, desde donde los axones se proyectan a regiones cerebrales involucradas en la regulación del ciclo sueño-vigilia, la memoria y los procesos que subyacen en la alimentación y la saciedad. El bloqueo de los receptores H1 en el cerebro explica el efecto sedante de los antihistamínicos «de primera generación», aquellos que atraviesan la barrera hemática cerebral.
En el año 1983, el grupo de trabajo dirigido por Jean-Charles Schwartz describió que la histamina inhibe su propia secreción en neuronas corticales aisladas de cerebro de rata. Estos experimentos condujeron al hallazgo de un tercer tipo de receptor para la histamina (además de los receptoresl H1 y H2 entonces conocidos). Se le designó como H3.
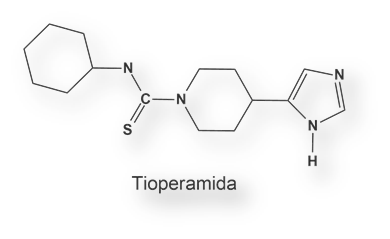
El grupo de Jean-Charles Schwartz sintetizó un agonista y un antagonista para el receptor H3: R (α) metilhistamina y tioperamida respectivamente. La activación del receptor H3 cerebral conduce a una ¯ AMPC y de la concentración de Ca2+ intracelular.
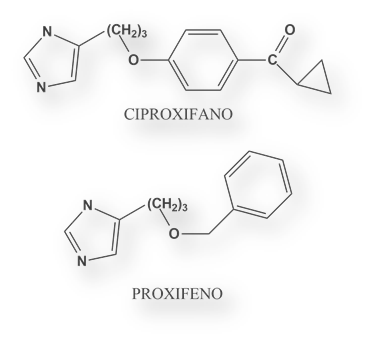
Otros antihistamínicos H3 son Ciproxifano y Proxifano.
Por otra parte, el equipo de trabajo de Manfred Göthert mostró que la activación de los receptores H3 inhibe la liberación de otros neurotransmisores tales como noradrenalina y serotonina. Así pues, la histamina, a través de la interacción con receptores H3, modula la liberación de sí misma (autorreceptores) y de otros neurotransmisores (heterorreceptores). Estos hallazgos han sido un acicate para la investigación de potenciales medicamentos que, interactuando con el receptor H3, puedan ser útiles en diversas enfermedades neurológicas y en el tratamiento de la obesidad.
EL RECEPTOR H4 DE LA HISTAMINA
Existe un cuarto tipo de receptor de la histamina: H4. [Referencia bibliográfica: Thurmond RL., et al. The role of histamine H1 and H4 receptors in allergic inflammation: the search for new antihistamines. Nat Rev Drug Discov 2008; 7: 41-53].
El receptor histaminérgico H4 se expresa en las células Th2 o linfocitos T CD4+, previamente estimulados por interleucina-4 (IL4). Este proceso está involucrado en la inflamación alérgica y en una de sus expresiones clínicas, el prurito. Aun cuando se sabe de la implicación de los receptores H4 tanto en procesos pro-inflamatorios como anti-inflamatorios, ambos imbricados en la inflamación alérgica, la intervención sobre estos receptores puede tener importantes aplicaciones farmacológicas.
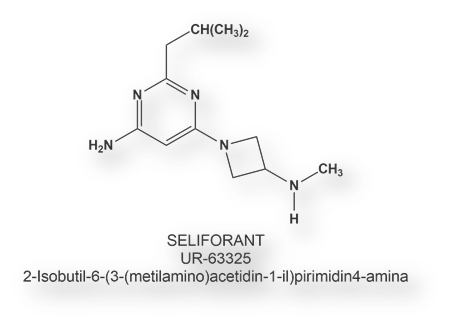 Un primer antagonista H4 (UR-63325 o Seliforant – International Non adopted Name o INN), sintetizado por el laboratorio Palau Pharma, se halla en estadio de ensayo clínico [Salcedo, C., et al Is the H4 receptor a new drug target for allergies and asthma? Front Biosci 2013; 5: 178-187]. Se está estudiando para tres indicaciones: desórdenes vestibulares, asma y rinitis alérgica.
Un primer antagonista H4 (UR-63325 o Seliforant – International Non adopted Name o INN), sintetizado por el laboratorio Palau Pharma, se halla en estadio de ensayo clínico [Salcedo, C., et al Is the H4 receptor a new drug target for allergies and asthma? Front Biosci 2013; 5: 178-187]. Se está estudiando para tres indicaciones: desórdenes vestibulares, asma y rinitis alérgica.
De otra parte, antagonistas mixtos H1/H4, o la combinación en una misma formulación farmacéutica de un antagonista H1 y otro antagonista H4, podrían pavimentar el futuro desarrollo de estrategias farmacéuticas novedosas para patologías inflamatorias e inmunológicas.
Además, los receptores H4 se hallan involucrados en la patogénesis de enfermedades no alérgicas. El bloqueo farmacológico de los receptores H4 disminuye la acumulación de neutrófilos en modelos experimentales de peritonitis y pleuresía.
Además, la activación de los receptores H4 induce la quimiotaxis de la interleucina-2 (IL-2) activada por las células NK (Natural Killers). De esta observación se infiere la posible utilidad del antagonismo del receptor H4 en una variedad de enfermedades, desde diabetes, cáncer, dolor neuropático y desórdenes vestibulares.
Zaragoza, a 4 de marzo de 2020
Dr. José Manuel López Tricas
Farmacéutico especialista Farmacia Hospitalaria
Farmacia Las Fuentes
Florentino Ballesteros, 11-13
50002 Zaragoza
[1] Black JW., et al. Definition and antagonism of histamine H2-receptors. Nature 1972; 236: 385-390.
[2] Jensen KG., et al. Cimetidine and gastric ulcer healing. Br Med J 1972; 2: 1479
[3] Bradshaw J., et al. Ranitidine (AH 19065): a new potent, selective histamine H2-receptor antagonist [proceedings] Br J Pharmacol 1979; 66: 464P.