El Organismo Regulador de Alimentos y Fármacos estadounidense (FDA, de su acrónimo en inglés, Food and Drug Administration) ha aprobado una terapia génica para la atrofia muscular espinal, denominada Zolgensma, indicada para niños de menos de 2 años afectados del «tipo 1» de la enfermedad (véase más adelante). El fabricante de esta terapia génica, la multinacional helvética Novartis AG., ha establecido un precio de venta de 2,1 millones de dólares para una única administración intravenosa. Es, con mucho, el tratamiento farmacológico más caro de cuantos existen hoy día. Las denominaciones de Zolgensma son: onasemnogene abeparvovec xioi, AAV9-CBA-SMN1-gene-therapy-AveXis; Adeno-associated-serotype-9-chicken-beta-actin-survival-motor-neuron-gene-therapy-AveXis; AVXS 101; y: ChariSMA™.
Se trata de un tratamiento que, al menos teóricamente, puede curar esta enfermedad hereditaria mortal.
Zolgensma es una terapia génica basada en adenovirus genéticamente modificados que actúan como transportadores de una copia corregida del gen deficitario en los niños con el tipo 1 de la atrofia muscular espinal. Una infusión intravenosa con estos adenovirus modificados genéticamente consigue que los genes se inserten en las neuronas motoras con la subsiguiente mejoría de la función motora del niño que, dada su corta edad, se manifiesta en su capacidad para erguir su cabeza, así como la de permanecer sentados.
Los efectos más frecuentes de la infusión de Zolgensma son la elevación de los niveles de enzimas hepáticas y los vómitos. Caso de existir una insuficiencia hepática, Zolgensma es propenso a desencadenar un cuadro hepático grave. Este escenario es poco probable dado que se administra a niños muy pequeños (menores de 2 años). No obstante, es imperativo un control protocolizado de las enzimas hepáticas durante al menos tres meses tras la infusión de Zolgensma.
Dado que Zolgensma está formado por adenovirus, se ha de valorar la idoneidad de postergar los programas de vacunación, sobre todo si se administran de manera concomitante tandas de corticoides.
Zolgensma tiene la consideración de medicamento huérfano. Su solicitud ha sido resuelta en base a la Fast Track, Breakthrough Therapy y a la Priority Review. Además se le otorgó prioridad al tratarse de una grave enfermedad pediátrica. AveXis, división de Novartis AG que desarrolló esta terapia génica, presentó la solicitud de autorización (Biologic License Application) en diciembre de 2018.
Algunas de las nuevas terapias génicas para algunas enfermedades raras cuestan cientos de miles de dólares; pocas, sin embargo, han traspasado 1 millón de dólares; y Zolgensma ha superado los 2 millones de dólares.
Por ejemplo, Luxturna®, una terapia génica para una forma de ceguera hereditaria, tiene un coste de $850,000; y Kymriah® (tisagenlecleucel), una terapia celular contra la leucemia linfoblástica aguda de células B para pacientes con edades no superiores a 25 años, tiene un precio de $475,000.
Luxturna® (voratigene neparvovec rzyl) es una versión corregida del gen RPE65. Este gen codifica una proteína retiniana fundamental para la transducción (conversión de la conformación trans del pigmento retiniano retinal en la conformación cis), proceso que permite la visión. Se usa para la distrofia retiniana asociada con una mutación (no necesariamente idéntica) en las dos copias (alelos) del gen RPE65. RPE65 es el acrónimo de Retinal Pigment Epithelium, proteína con un peso molecular de 65 Kd.
Novartis AG., ha presentado a las compañías aseguradoras un plan de financiación de Zolgensma consistente en pagar $425,000 anuales durante un lustro.
Novartis se había negado hasta ahora a hacer público el precio de Zolgensma, mientras sus ejecutivos se han prodigado en medios de comunicación para preparar a la comunidad médica y la sociedad ante el precio de esta terapia génica.
En uno de los encuentros, un ejecutivo de Novartis afirmó ante la prensa que el laboratorio comercializaría el tratamiento a un coste mucho más bajo del inicialmente previsto, de entre 4 y 5 millones de dólares.
David Lennon, presidente de AveXis, la división de Novartis que desarrolló Zolgensma declaró que la compañía farmacéutica fijó un precio 50% inferior a lo que inicialmente tenía previsto, hallándose en el rango de precios de otras terapias para enfermedades raras.
Otro medicamento para la atrofia muscular espinal, Nusinersén, registrado como Spinraza® fue autorizado en el año 2016. El primer año de tratamiento exige un desembolso de $750,000; y $375,000 anualmente a partir del segundo año. Así pues, el coste para una década supera los 4 millones de dólares.
Los ejecutivos de Novartis declaran que Zolgensma puede otorgar a niños con una enfermedad mortal la posibilidad de llevar una existencia libre de las gravísimas limitaciones físicas asociadas con la atrofia muscular espinal.
La atrofia muscular espinal afecta a la placa motora, la sinapsis que conecta a las neuronas motoras con los músculos esqueléticos que inervan. El gen responsable de la atrofia muscular espinal se localizó en el brazo corto (q) del cromosoma 5 (5q) en el año 1992. Tres años más tarde, un grupo de investigación dirigido por Judith Melki identificó la proteína codificada por dicho gen. La proteína se designa como SMN, acrónimo en inglés de Survival Motor Neuron. Como se infiere de su denominación, SMN es fundamental para el funcionamiento correcto de las neuronas motoras y los músculos esqueléticos inervados por aquéllas.
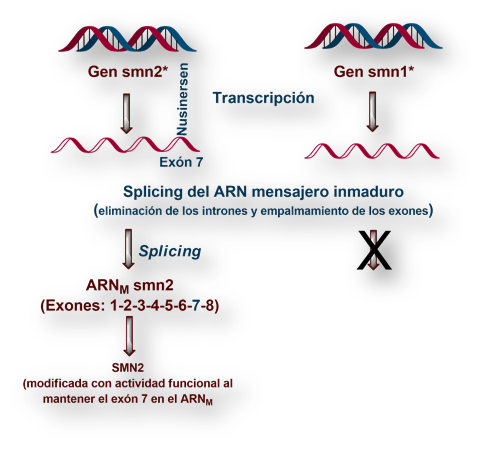 Cada uno de las dos copias (alelos) del gen de la atrofia muscular espinal contiene información codificada para la síntesis de una de las dos versiones de SMN, esto es, SMN1 y SMN2 respectivamente.
Cada uno de las dos copias (alelos) del gen de la atrofia muscular espinal contiene información codificada para la síntesis de una de las dos versiones de SMN, esto es, SMN1 y SMN2 respectivamente.
Mientras SMN1 es una proteína plenamente funcional, el ARN mensajero de SMN2 (carente del exón 7) se traduce en una versión incompleta de la proteína. La función del ARN m de SMN2 parece consistir en modular la expresión del gen que codifica SMN1.
La diferencia entre SMN1 y SMN2 radica en el exón 7, específicamente en el primer nucleótido, citosina en el gen que codifica SMN1; y timina en el gen que codifica SMN2. Tras el proceso de maduración (splicing), el ARN m de SMN1 contiene el exón 7, mientras el SMN2 carece de dicho exón.
El alelo que codifica la síntesis de SMN2, del que existen varias copias, actúa como modulador de la expresión del alelo para la síntesis de SMN1. Cuando el alelo de SMN1 ha sufrido una mutación, el número de copias que se sintetizan de SMN2 determina la gravedad de la sintomatología de la atrofia muscular espinal (cuanto mayor número de copias de SMN2, menor gravedad, indicativo de que proteína SMN1 retiene mayor actividad funcional).
Aun cuando la proteína SMN2 es solo parcialmente funcional, puede contribuir a suplir la falta de la proteína completa SMN1. De alguna manera, el alelo para SMN2 se puede considerar un gen de respaldo, en caso de deficiencia de la proteína funcional codificada por el gen SMN1.
Bajo el sintagma atrofia muscular espinal se engloban un conjunto de genopatías musculares que, en conjunto, representan la segunda causa de enfermedad neurodegenerativa, siendo la primera la distrofia muscular de Duchenne. La prevalencia de la atrofia muscular espinal es de 1 caso por cada 100.000 nacimientos vivos.
La atrofia muscular espinal se clasifica en cinco tipos:
La atrofia muscular espinal tipo 1, también denominada enfermedad de Werdnig-Hoffmann, debuta alrededor de los 6 meses de edad. Cursa con hipotonía, fasciculaciones, y dificultad para deglutir y respirar. Los niños afectados no llegan a gatear, sentarse o permanecer erguidos. Tristemente suelen fallecer antes de su segundo aniversario.
La atrofia muscular espinal tipo 2 se manifiesta entre los 6 y 18 meses de edad. Los niños consiguen sentarse por sí mismos, pero son incapaces de mantenerse erguidos sin ayuda de terceros. Las infecciones son comunes. Su esperanza de vida no suele sobrepasar la adolescencia.
La atrofia muscular espinal tipo 3, llamada también enfermedad de Kugelberg-Welander, debuta entre los 2 y los 17 años. Los niños tienen una marcha anormal, dificultad para correr, levantarse de una silla, y temblor fino de los dedos. Además, padecen escoliosis (a semejanza de los niños con el «tipo 1»), y contracturas debidas a acortamiento de músculos o tendones peri-articulares. La frecuencia de infecciones respiratorias es elevada en relación a su grupo de edad y sexo. Su morbilidad es superior a la media, pero su mortalidad similar a la de la población general.
La atrofia muscular espinal con artrogriposis es una versión muy infrecuente. Las manifestaciones clínicas incluyen contracturas graves, escoliosis, deformidad del tórax, mandíbulas muy poco desarrolladas y ptosis parpebral.
La atrofia muscular espino-bulbar progresiva o enfermedad de Kennedy, puede aparecer en algún momento entre aproximadamente 15 años y la sexta década de la vida. Se manifiesta clínicamente con debilidad de los músculos de la cara, mandíbula y lengua, que trasunta en problemas de masticación, deglución y dicción. La progresión de la atrofia da lugar a fasciculaciones y alteraciones sensoriales en manos y pies debido a neuropatía sensorial (degeneración nerviosa sensorial).
La consecuencia clínica de la atrofia muscular espinal en su versión más grave («tipo 1») es la pérdida de fuerza física hasta imposibilitar la deambulación, deglución; así como la capacidad de articular sonidos y finalmente respirar de manera autónoma.
Antes de la comercialización de Spinraza® (Nusinersen) los niños con el «tipo 1» de la enfermedad fallecían antes de cumplir 2 años.
Aproximadamente 1 de cada 11.000 niños nacidos vivos sufren atrofia medular espinal.
La Food and Drug Administration (FDA) estadounidense ha aprobado Zolgensma para todas las formas de atrofia muscular espinal en niños de menos de 2 años de edad.
Se estima que solo en Estados Unidos se diagnostican alrededor de 400 nuevos casos al año, unos 30 cada mes. Existen otros 700 aproximadamente susceptibles de beneficiarse del tratamiento.
En ensayo clínico (STR1VE) en el que se apoyó la Food and Drug Administration para la aprobación de Zolgensma incluyó solo a 36 niños. Sin embargo, Novartis AG., afirmó haber usado esta terapia génica en aproximadamente 150 niños. El ensayo clínico STR1VE se denominó Gene Replacement Therapy Clinical Trial for Patients With Spinal Muscular Atrophy type 1.
Algunos niños tratados antes de los dos años se hallan ahora en su quinto cumpleaños sin merma de la mejoría obtenida inicialmente. No solo los padres, sino los científicos, ejecutivos del laboratorio, y gestores de recursos sanitarios observan su evolución con enorme interés.
Hay un problema añadido a la asunción del coste del tratamiento con Zolgensma: es si los niños que actualmente reciben Nusinersen (Spinraza®) se derivarán al tratamiento con Zolgensma.
El coste del tratamiento está fuera del alcance de prácticamente todas las familias. Asumiendo que la factura será pagada por las compañías aseguradoras o, si se aprueba en otros países, por su seguridad social, Novartis ha declarado que llevaría a cabo sustanciales descuentos si el niño fallece o no consigue una respuesta suficiente (por ejemplo, termina precisando respiración asistida) Todas estas cuestiones se han de resolver rápidamente, porque la eficacia del tratamiento está condicionada a que éste se lleve a cabo enseguida tras el diagnóstico confirmatorio.
Zolgensma se encuadra en las terapias génicas. Se trata de una tecnología novedosa, pero carente de prospectiva, por lo que no se puede prever que la mejoría lograda se prolongue a lo largo de la vida.
La respuesta ante Zolgensma es fundamental porque durante el próximo lustro se prevén alrededor de 60 nuevas terapias génicas para enfermedades raras, todas ellas a precios muy elevados (datos de EvaluatePharma).
Solo las naciones con economías muy solventes y sistemas de aseguramiento sanitario muy consolidado pueden asumir los costes de estos tratamientos. ¿Qué sucederá con los recién nacidos afectados de atrofia muscular espinal tipo 1 en otros países?
Zaragoza, a 31 de mayo de 2019
Dr. José Manuel López Tricas
Farmacéutico especialista Farmacia Hospitalaria
Farmacia Las Fuentes
Florentino Ballesteros, 11-13
50002 Zaragoza