
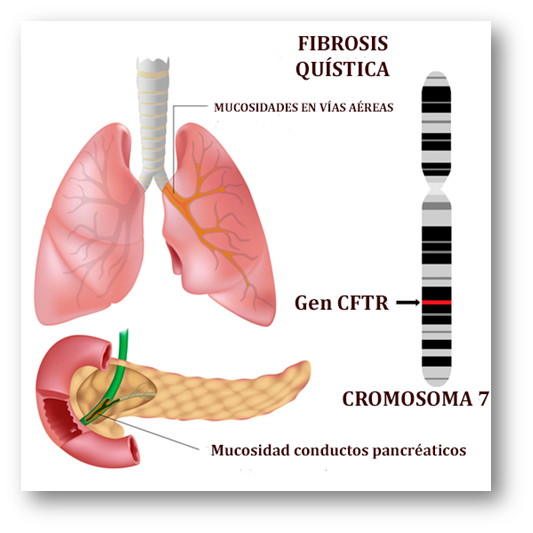
La fibrosis quística (mucoviscidosis) es una enfermedad genética autosómica recesiva caracterizada por la acumulación de mucosidades en conductos de diversos órganos corporales. Los más afectados son los pulmones (mucosidades en bronquios y bronquiolos), páncreas (mucosidades en conductos pancreático y biliar), glándulas sudoríparas, y sistema reproductor (mucosidad en los conductos deferentes). La sintomatología deriva principalmente de la afectación de los órganos citados.
De sólito, los revestimientos de los conductos corporales se hallan recubiertos de una sustancia viscosa (moco) que los lubrica y protege. Sin embargo, en los pacientes de fibrosis quística, este moco (viscoso y pegajoso) se halla en cantidades excesivas debido a la imposibilidad de un drenaje fisiológico. En estas circunstancias, la función del órgano afectado resulta comprometida. Además, la mucosidad constituye un caldo de cultivo para el crecimiento bacteriano. La reacción fisiológica es la inflamación y subsiguiente desarrollo de tejido cicatricial (fibrosis) que empeora la prognosis de la enfermedad.
 Aun cuando el deterioro de la función pulmonar es el signo más grave de la fibrosis quística, la afectación digestiva también es importante. Algunos recién nacidos con fibrosis quística sufren tras el parto, antes del meconio (primera defecación) un bloqueo intestinal.
Aun cuando el deterioro de la función pulmonar es el signo más grave de la fibrosis quística, la afectación digestiva también es importante. Algunos recién nacidos con fibrosis quística sufren tras el parto, antes del meconio (primera defecación) un bloqueo intestinal.
Uno de los órganos intestinales más perjudicados es el páncreas, comprometiendo la secreción de enzimas digestivas necesarias para la digestión y procesamiento de los nutrientes. Secundariamente, el deterioro de la función pancreática dificulta la secreción de insulina, causando una forma indirecta de diabetes. Otros síntomas digestivos incluyen: diarrea, desnutrición, retardo del crecimiento y emaciación.
Hasta no hace muchos años los niños con fibrosis quística solían fallecer durante la primera infancia. Hoy día, la mayoría de los pacientes llegan a la adultez, controlando la sintomatología asociada.
La acumulación de mucosidades en el sistema reproductor causa infertilidad.
Un editorial publicado en la revista New England Journal of Medcine da cuenta de dos estudios clínicos (publicados en New England Journal of Medicine, y The Lancet) que informan de los resultados favorables de una «triple terapia» para la fibrosis quística derivada de una mutación (Phe508) en el gen que codifica la síntesis de la proteína de membrana designada con el acrónimo CFTR (Cystic Fibrosis Trans-membrane Conductance Regulator). Estos estudios señalan que pronto será posible ofrecer terapias seguras y eficaces para el 90% aproximadamente de los enfermos. El editorial está firmado por Francis S. Collins, quien en el año 1989, conjuntamente con Lap-Chee-Tsu, identificaron el gen responsable de esta genopatía.
El gen, designado Phe508, se sitúa en el brazo largo del cromosoma 7. [Phe508 hace referencia a que la mutación afecta al codón (triplete nucleótido) que inserta el aminoácido fenilalanina (Phe) en posición 508 de la secuencia de aminoácidos de la proteína CFTR. La genopatía manifiesta toda su clínica en portadores homocigóticos del gen mutado, a lo largo del primer año de vida, durante el período de lactancia.
CFTR es una proteína transmembrana que forma parte esencial del canal iónico para transporte del anión cloruro. Como tal, es fundamental para un balance correcto de sal y agua en los pulmones, páncreas, glándulas sudoríparas y otros órganos. De hecho, el sudor de los niños con fibrosis quística es salado, circunstancia que permitió desarrollar el «test del sudor» para el diagnóstico de la enfermedad.
La mutación Phe508 del gen CFTR es la más común de las aproximadamente 1.700 mutaciones del gen identificadas hasta ahora, todas ellas vinculadas con el desarrollo de fibrosis quística. La proteína CFTR errónea, al tener una conformación defectuosa, queda atrapada en el retículo endoplásmico; y, además, las moléculas que finalmente consiguen insertarse en la membrana celular no pueden ser activadas por su ligando fisiológico. Por lo tanto, la estrategia farmacológica debe ir dirigida tanto a corregir el plegamiento erróneo con fármacos «correctores», como a posibilitar la activación de la proteína embebida en la membrana con medicamentos «potenciadores». CFTR es una proteína compleja; su estructura y función requirió una década de investigación.
La «joint venture» de la Cystic Fibrosis Foundation y un pequeño laboratorio, Aurora BioSciences (más tarde denominado Vertex Pharmacéuticals) permitió durante la década de 1990 iniciar un extenso programa de investigación dirigido a buscar medicamentos «correctores» [del plegamiento erróneo de CFTR] y «potenciadores» [de la CFTR ya insertada en la membrana celular]. Las estrategias urdidas en este programa de investigación resultaron de gran interés en la investigación de otras enfermedades genéticas.
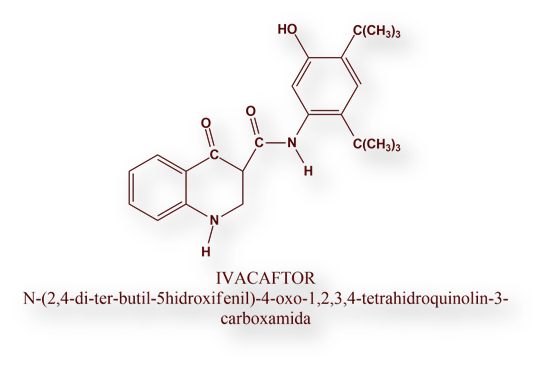
La Food and Drug Administration (FDA) estadounidense autorizó en el año 2012 el primer fármaco para un tipo de mutación muy infrecuente en la fibrosis quística. Se trató de Ivacaftor (Kalydeco®) dirigido contra la mutación G551D del gen CFTR, que afecta a un 5% aproximadamente de todos los pacientes con fibrosis quística Esta mutación codifica una proteína CFTR que, si bien adquiere la conformación correcta en el retículo endoplásmico, no puede ser activada en su ubicación en la membrana celular. Se trata, pues, de un fármaco «activador».
En el año 2015, la Food and Drug Administration autorizó Orkambi®, asociación de Ivacaftor y Lumacaftor.
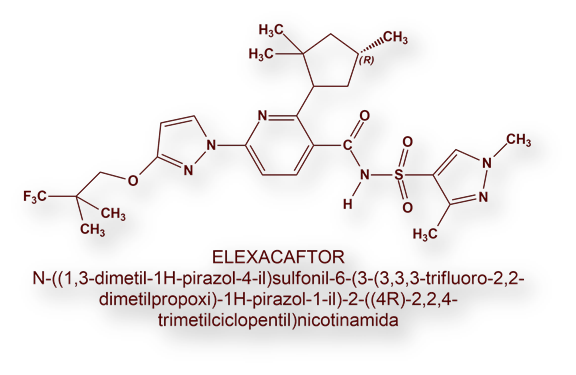
En 2018, la US Food and Drug Administration autorizó la asociación «Tezacafator-Ivacaftor» para la mutación Phe508 del gen CFTR en pacientes con dos alelos mutados, o un solo alelo afectado de otras 26 mutaciones; y en 2019 aprobó la «triple terapia» («Elexacaftor-Tezacaftor-Ivacaftor») para la mutación Phe508 del gen CFTR en pacientes homocigotos o heterocigotos. Mientras Ivacaftor es un «potenciador», Elexacaftor y Tezacaftor son «correctores».
La fibrosis quística ocurre en 1 de cada 2.500 a 3.500 niños caucasianos. Su prevalencia es menor en otros grupos étnicos: 1 de cada 17.000 afroamericanos; y 1 de cada 31.000 asiáticos. En países como Dinamarca, más del 87% de la población son portadores de la mutación Phe508, mientras en otros como Turquía la prevalencia de dicha mutación es inferior al 21%.
La enfermedad solo se manifiesta clínicamente en los homocigotos (las dos copias del gen – alelos –) mutados. En los heterocigotos (un alelo mutado, el otro normal) la enfermedad no se aparece o su clínica el leve.
Hasta no hace muchos años los niños afectados de fibrosis quística fallecían durante sus primeros años de vida. En la actualidad la mitad aproximadamente de los afectados alcanzan la adultez.
La «triple terapia» (Trikafta®) asocia los siguientes fármacos: Elexacaftor, Tezacaftor e Ivacaftor.

 Los pacientes tratados con esta «triple terapia» mostraron mejoría de los parámetros de función pulmonar en los dos estudios clínicos referidos.
Los pacientes tratados con esta «triple terapia» mostraron mejoría de los parámetros de función pulmonar en los dos estudios clínicos referidos.
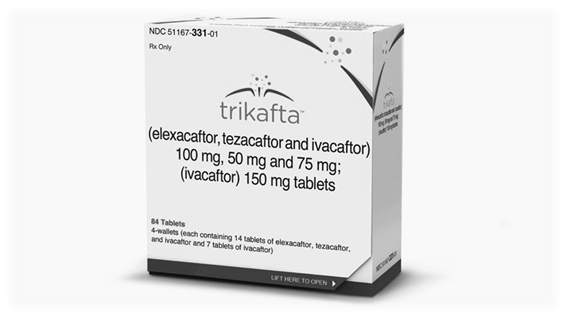 Los ensayos se realizaron gracias a la financiación del fabricante de Trikafta®, Vertex Pharmaceuticals. La Food and Drug Administration (FDA) estadounidense autorizó el uso de Trikafta® en pacientes con edades iguales o superiores a 12 años portadores de la mutación Phe508 del gen CFTR, la más común, de todas las causantes de fibrosis quística.
Los ensayos se realizaron gracias a la financiación del fabricante de Trikafta®, Vertex Pharmaceuticals. La Food and Drug Administration (FDA) estadounidense autorizó el uso de Trikafta® en pacientes con edades iguales o superiores a 12 años portadores de la mutación Phe508 del gen CFTR, la más común, de todas las causantes de fibrosis quística.
El coste del tratamiento anual será (en Estados Unidos) de $331,000 (tres cientos once mil dólares), un precio similar al medicamento ya en uso para esta enfermedad, Kakydeco® (Ivacaftor). La empresa farmacéutica se compromete a implementar programas que faciliten el acceso a todos los pacientes que lo precisen.
Vertex Pharmaceuticals ha sido objeto de críticas por los elevados precios de sus medicamentos para la fibrosis quística. En un informe (2018 Report) del Institute for Clinical and Economic Review se incide en que estos fármacos deberían costar un 77% de su precio actual.
El «talón de Aquiles» de esta terapia se halla en el grupo de pacientes, aproximadamente un 5%, para los que no existen hoy día opciones terapéuticas.
Zaragoza, a 12 de noviembre de 2019
Dr. José Manuel López Tricas
Farmacéutico especialista Farmacia Hospitalaria
Farmacia Las Fuentes
Zaragoza