La «deficiencia de ornitina-transcarbamilasa» es una genopatía que da lugar a hiperamonemia. Las elevadas concentraciones plasmáticas de ion amonio (la forma biológicamente activa del amoníaco) son especialmente tóxicas para el cerebro.
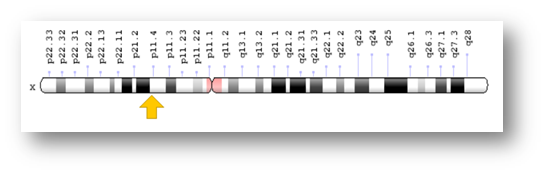
La «deficiencia de ornitina-transcarbamilasa» afecta a los niños varones, siendo muy rara en las niñas (el gen que codifica la síntesis de la enzima ornitina-transcarbamilasa se ubica en el cromosoma X). Desde un punto de vista citogenético es Xp11.4, significando que se halla en el brazo corto (q) del cromosoma X en la posición 11.4 respecto del centrómero).
En su versión más grave la «deficiencia de ornitina-transcarbamilasa» debuta el primer o segundo día de vida del recién nacido. El niño permanece letárgico, incapaz de succionar la leche materna (o el sucedáneo adaptado), con muy escasa regulación de su temperatura corporal y su respiración, crisis convulsivas, derivando hacia un estado comatoso. Además sufre una grave y progresiva hepatopatía.
En algunos afectados la sintomatología es mucho más leve y debuta en la edad adulta tardía. Esta versión de la deficiencia tiene idéntica prevalencia en hombres y mujeres. Algunas manifestaciones clínicas de esta forma tardía de la «deficiencia de ornitina transcarbamilasa» incluyen conducta errática, disminución del nivel de conciencia, cefaleas, vómitos, aversión a alimentos proteicos, y convulsiones.
Los afectados de «deficiencia de ornitina-transcarbamilasa» en su manifestación más grave (recién nacidos) carecen del gen que codifica la síntesis de esta enzima. En cambio la presentación más tardía y con sintomatología auto-limitada se debe a la presencia de un gen defectuoso pero con actividad residual.
La ornitina-transcarbamilasa es la primera enzima del «ciclo de la urea». La ausencia total de esta enzima, o la síntesis de un enzima con baja o muy baja actividad, bloquea el «ciclo de la urea». La consecuencia es la incapacidad de eliminar el nitrógeno. Somos animales ureotélicos (véase más adelante en este mismo informe).
La síntesis de urea (carbamida) en el hígado es la ruta principal de eliminación de iones amonio (NH4+).
Un bloqueo de la síntesis de carbamoilfosfato (carbamilfosfato) o de cualquiera de las cuatro etapas del «ciclo de la urea» causa efectos devastadores porque no existe una ruta alternativa para la biosíntesis de urea. [Recuérdese que el «ciclo de la urea» o «ciclo de carbamida» fue la primera vía metabólica cíclica que se desentraño, en el año 1932, hito de la bioquímica debido a Hans Adolf Krebs].
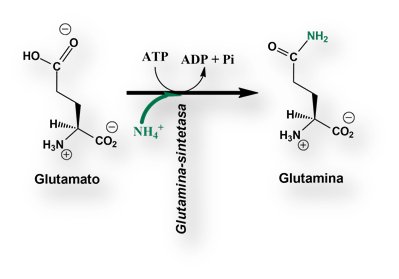 Todos los defectos metabólicos del «ciclo de la urea» conducen a un incremento de las concentraciones de ion amonio en sangre (hiperamonemia). ¿Por qué la hiperamonemia es tan tóxica? La cuestión no se ha dilucidado, si bien parece que las concentraciones elevadas de glutamato y glutamina provocan efectos osmóticos que desencadenan gravísimos daños cerebrales.
Todos los defectos metabólicos del «ciclo de la urea» conducen a un incremento de las concentraciones de ion amonio en sangre (hiperamonemia). ¿Por qué la hiperamonemia es tan tóxica? La cuestión no se ha dilucidado, si bien parece que las concentraciones elevadas de glutamato y glutamina provocan efectos osmóticos que desencadenan gravísimos daños cerebrales.
Una de las genopatías relacionadas con la bioquímica de la urea es la deficiencia de alguna de las dos enzimas siguientes: carbamilfosfato-sintetasa; y la ornitina-transcarbamilasa. Una estrategia para soslayar este error del metabolismo es la siguiente:
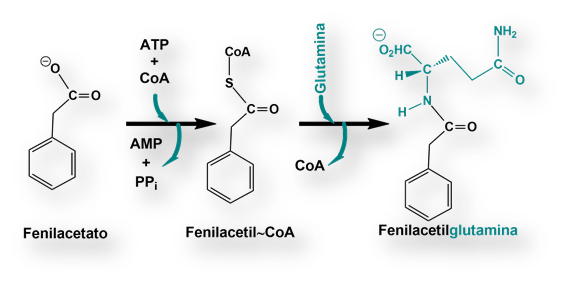
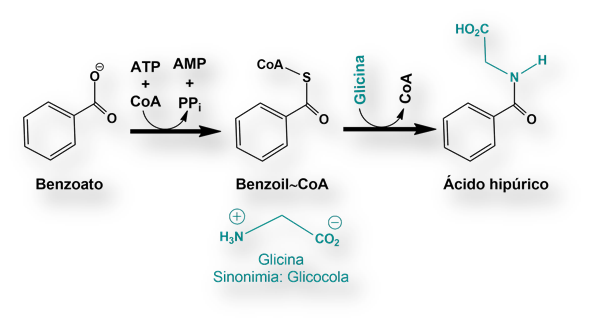
Los niños con «deficiencia de la ornitina-transcarbamilasa», no pueden usar la citrulina y el arginina-succinato para deshacerse del nitrógeno. La síntesis de estos dos intermediarios metabólicos se halla bloqueada. Bajo estas circunstancias se acumula nitrógeno en forma de glicina y glutamina. El objetivo es, pues, eliminar estos dos aminoácidos del organismo. Para ello se aporta una dieta pobre en proteínas pero suplementada con grandes cantidades de benzoato y fenilacetato (a veces, apocopada como fenacetato). Ambos compuestos se convierten en hipurato y fenilacetil-glutamina respectivamente, que se excretan por vía renal.
La mayoría de los organismos terrestres son ureotélicos, esto es, excretan el excedente de nitrógeno en forma de urea.
Sin embargo, la urea no es la única vía de eliminación del nitrógeno. Los vertebrados e invertebrados acuáticos eliminan el exceso de nitrógeno directamente en forma de ion amonio (NH4+), denominándose por ello animales amoniotélicos.
La evolución ha logrado éxitos impresionantes. Así, por ejemplo, el llamado pez pulmonado, de sólito amoniotélico (excreta ion amonio), se convierte en uretotélico (excreta urea) cuando las condiciones de su hábitat se tornan desfavorables, durante los períodos de sequía en los que se ve obligado a vivir fuera del agua.
Tanto los animales ureotélicos como los amoniotélicos precisan de suficiente cantidad de agua para la excreción del excedente de nitrógeno, bien en forma de urea (ureotélicos), bien como catión amonio (amoniotélicos).
Los organismos uricotélicos excretan el exceso de nitrógeno en forma de ácido úrico (una base púrica). Esta ruta de eliminación es muy poco exigente en agua. Esta forma de eliminación (como ácido úrico) es una adaptación evolutiva muy útil en animales que ponen huevos, con membranas impermeables que acumulan productos de desecho. Así, pues, la ruta de eliminación del nitrógeno ha evolucionado en función del hábitat, y sus condiciones de hidratación.
Zaragoza a 20 de octubre de 2017
Dr. José Manuel López Tricas
Farmacéutico especialista Farmacia Hospitalaria
Farmacia Las Fuentes
Zaragoza