VARICELA. CONCEPTOS BÁSICOS
La varicela es una infección aguda causada por el virus varicela zóster (abreviadamente ZVZ). La infección recurrente se denomina herpes zóster.
Hasta el siglo XIX la varicela y la viruela se consideraban la misma enfermedad. Steiner demostró en el año 1875 que la varicela es una enfermedad infecciosa: la inoculación a personas sanas del fluido de las vesículas de un enfermo transmitía la enfermedad. [La viruela es una enfermedad erradicada desde la década de 1970, conservándose algunas cepas en unos pocos laboratorios de alta seguridad].
En el año 1888 experimentos realizados por von Bokay demostraron la relación entre la varicela y el herpes zóster.
Muchos años después, en 1954, Thomas Weller aisló un mismo tipo de virus, tanto de las vesículas de pacientes con varicela como de ampollas de pacientes con herpes zóster. No había dudas: se trataba de dos manifestaciones clínicas del mismo proceso infeccioso.
La primera infección por el virus ZVZ da lugar a varicela. Tras la resolución del proceso infeccioso, el virus no se elimina, sino que se acantona en estado latente en los ganglios del sistema nervioso sensorial. La reactivación del virus, muchos años después, da lugar a un cuadro patológico denominado herpes-zóster, o simplemente herpes. En cualquier caso se trata del mismo virus.
[Los ganglios nerviosos constituyen grupos neuronales ubicados fuera del Sistema Nervioso Central. Éstos pueden ser: ganglios raquídeos (a ambos lados de la médula espinal), ganglios craneanos (adjuntos al encéfalo), y ganglios neurovegetativos (sitos al lado o en el interior de las vísceras].
 A comienzos de la década de 1970 se desarrolló en Japón la primera vacuna contra la varicela usando como antígenos virus con virulencia atenuada. La Food and Drug Administration (FDA) norteamericana autorizó esta vacuna (Varivax®) en el año 1995; y otra vacuna para reducir el riesgo de herpes zóster (Shingrix®) se aprobó en mayo de 2006, comercializada por GlaxoSmithKline Pharma, e indicada en personas de 60 o más años, sobre todo porque la protección contra el herpes-zóster se disipa con el tiempo.
A comienzos de la década de 1970 se desarrolló en Japón la primera vacuna contra la varicela usando como antígenos virus con virulencia atenuada. La Food and Drug Administration (FDA) norteamericana autorizó esta vacuna (Varivax®) en el año 1995; y otra vacuna para reducir el riesgo de herpes zóster (Shingrix®) se aprobó en mayo de 2006, comercializada por GlaxoSmithKline Pharma, e indicada en personas de 60 o más años, sobre todo porque la protección contra el herpes-zóster se disipa con el tiempo.
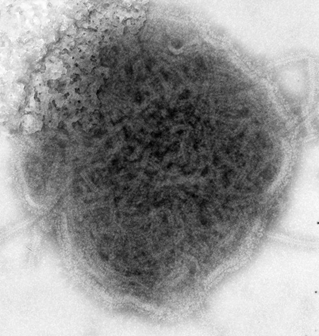
El virus varicela zóster es un ADN-virus perteneciente al grupo de los herpes-virus.
El virus de la varicela penetra en el organismo a través del tracto respiratorio y la conjuntiva. La replicación viral se produce en los ganglios linfáticos de la nasofaringe. Tras la infección se produce viremia a los 4 a 6 días, difundiendo desde la sangre a todos los órganos, con especial afinidad por el hígado, bazo y ganglios de los nervios sensoriales.
Tras su replicación en las vísceras se produce una segunda viremia, seguida de una infección de la piel. El virus se puede cultivar en células mononucleares del paciente desde 5 días antes y hasta 1 o 2 días después de la aparición de la dermatitis (ver epígrafe siguiente).
El período de incubación es de 14 a 16 días (rango: 10 a 21 días) tras la exposición al virus de la varicela o herpes zóster. El pródromo incluye fiebre, malestar; y, 1 o 2 días después, eritema, sobre todo en adultos. En cambio el eritema es el primer signo clínico en niños.
PATOGNOMONIA EN PERSONAS NO VACUNADAS
Se presenta eritema pruriginoso que progresa rápidamente a máculas y pápulas (eritema maculopapular). El eritema aparece primero sobre el rostro, sigue por el tórax, espalda, extendiéndose al resto del cuerpo. Las lesiones son predominantes en tórax y espalda.
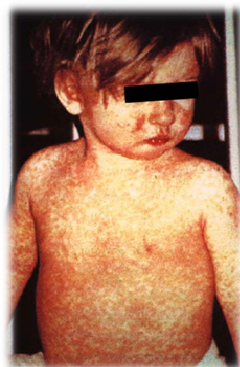
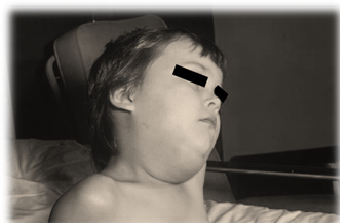
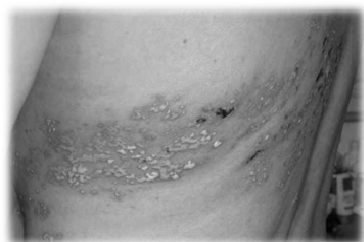 La enfermedad no es grave, excepto en pacientes con compromiso inmunológico. La infección otorga inmunidad de por vida. En pacientes con déficits inmunológicos se puede producir una reinfección por el virus de la varicela. Esta segunda reinfección no se denomina varicela, sino herpes zóster ya que su clínica es distinta (ver fotografía adjunta de paciente con herpes zóster).
La enfermedad no es grave, excepto en pacientes con compromiso inmunológico. La infección otorga inmunidad de por vida. En pacientes con déficits inmunológicos se puede producir una reinfección por el virus de la varicela. Esta segunda reinfección no se denomina varicela, sino herpes zóster ya que su clínica es distinta (ver fotografía adjunta de paciente con herpes zóster).
PATOGENIA EN PERSONAS VACUNADAS
La infección por el serotipo salvaje del virus varicela zóster en personas vacunadas ocurre 42 días después de la vacunación. El proceso es leve, con febrícula y pocas lesiones dérmicas (<50). El eritema no es vesicular. Sin embargo, entre el 25% y el 30% de las personas vacunadas sufren una infección con las mismas características que las no vacunadas.
El diagnóstico clínico es difícil, precisándose confirmación de laboratorio.
COMPLICACIONES DE LA VARICELA
Las más habituales son de dos tipos:
- Celulitis (infecciones de piel y tejidos blandos)
- Neumonía (solo en adultos)
Otras complicaciones graves, muy infrecuentes, incluyen ataxia cerebelar, encefalitis, hemorragias, septicemia tóxica, shock séptico, fascitis necrotizante, osteomielitis, neumonía (viral o bacteriana), y artritis séptica.
CONTAGIO DE LA VARICELA
La varicela es muy contagiosa, bien por inhalación de las secreciones expelidas por un enfermo, bien por contacto con sus lesiones vesiculares. El paciente es contagioso desde 1 o 2 días antes de que debute el eritema hasta que las vesículas han formado costra (de 10 a 21 días tras el inicio de la infección). Se considera que el 90% de las personas no vacunadas que contacten con un enfermo de varicela contraerán la infección.
La varicela es menos contagiosa que el sarampión, pero más que la parotiditis y la rubéola.
RIESGOS DE LA VARICELA EN EMBARAZADAS
Las embarazadas que se contagian con el virus de la varicela pueden desarrollar una neumonía potencialmente mortal. El riesgo de neumonía y la probabilidad de graves complicaciones parece ser mayor durante el tercer trimestre del embarazo, si bien no hay consenso en este aspecto.
Si la gestante contrae varicela durante el primer o segundo trimestre de su embarazo, el riesgo de que el recién nacido sufra el «Síndrome de varicela congénita» es relativamente bajo (0,4% a 2%). El recién nacido puede tener bajo peso y deformidades en cerebro, ojos y miembros.
Si la gestante desarrolla dermatitis por varicela desde 5 días antes a 2 días después del parto, el recién nacido tiene un riesgo importante (aproximadamente 30%) de sufrir varicela neonatal. Este porcentaje se reduce hasta el 7% si se actúa con determinación, administrando gammaglobulinas anti-varicela.
Está contraindicada de modo absoluto la administración de la vacuna contra la varicela en mujeres embarazadas (Guidelines for Vaccinating Pregnant Women: Varicella). Consultar también la página web Chickenpox and Pregnancy.
A pesar de disponer de una vacuna efectiva (dos dosis ofrecen una protección > 90%), todavía se siguen produciendo casos de varicela. En Estados Unidos la incidencia de varicela es de unos 4 millones de infecciones, de las que 10.600 precisan ingreso hospitalario, con una mortandad de 100 a 150 personas cada año.
La vacunación generalizada no solo es útil para la protección individual, sino también para disminuir la prevalencia de la infección. Es importante por cuánto hay personas que por sus especiales circunstancias (inmunodeprimidos, embarazadas) no pueden ser vacunadas, y deben beneficiarse de la protección derivada de la baja prevalencia del virus en la comunidad derivada de la vacunación masiva.
Zaragoza, a 16 de noviembre de 2017
Dr. José Manuel López Tricas
Farmacéutico especialista Farmacia Hospitalaria
Farmacia Las Fuentes
Zaragoza