
Las mitocondrias son orgánulos ovalados de 2 x 5 mcm (micrómetros), aproximadamente el tamaño de una bacteria típica. [La nomenclatura de micrómetro (10-6 metros) ha cambiado de «μm» a «mcm»].
Han transcurrido alrededor de 80 años desde que Eugene Kennedy y Albert Lehninger descubrieron los trascendentes procesos bioquímicos que se desarrollan en las mitocondrias: la fosforilación oxidativa o «cadena respiratoria», el «ciclo del ácido cítrico» (ciclo de Hans Krebs) y la β-oxidación de los ácidos grasos.
George Palade y Fritjof Sjöstrand, mediante microscopio electrónico, mostraron que las mitocondrias poseen dos membranas, una externa, y otra interna con numerosos pliegues. Esta disposición crea dos compartimentos: un espacio inter-membranoso (entre las membranas externa e interna); y una matriz mitocondrial, delimitada por la membrana interna.
El ciclo de Krebs («ciclo del ácido cítrico» o «ciclo de los ácidos tricarboxílicos», son otras denominaciones usuales) y la β-oxidación de los ácidos grasos se producen en la matriz mitocondrial; la «cadena respiratoria» (fosforilación oxidativa) en el compartimento inter-membranoso.
La membrana externa es permeable para iones y moléculas de pequeño peso molecular. Ello se debe a la abundancia de una proteína con un peso molecular entre 30 y 35 kd, denominada porina mitocondrial, más conocida por su acrónimo VDAC (Voltage Depend Anionic Channel). La configuración de VDAC remeda a las porinas bacterianas. [kd, acrónimo de kilo-dalton o quilo-dalton, siendo 1 dalton 1 unidad de masa atómica].
A diferencia de la membrana mitocondrial externa, la membrana mitocondrial interna es impermeable a iones y moléculas con polaridad. Por esta razón, el trasiego de moléculas se realiza mediante transportadores específicos. De éstos se benefician moléculas como ATP, piruvato y citrato.
Además, existe una diferencia de potencial, de tal suerte que la matriz mitocondrial es electronegativa, mientras la membrana externa, al igual que el citosol, electropositiva.
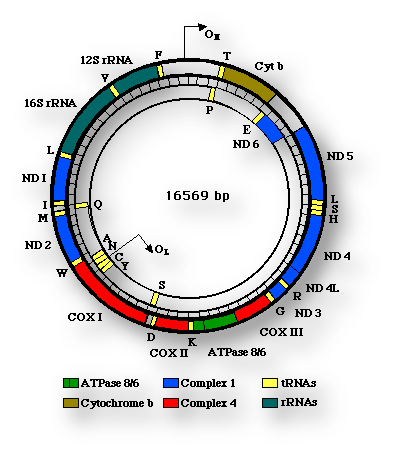 Las mitocondrias son orgánulos semiautónomos que mantienen una relación endosimbiótica con el «hospedador» (la propia célula). No en vano, estos orgánulos tienen su propio ADN, de un tamaño muy variable según las especies. El ADN mitocondrial humano tiene 16.569 pares de bases (indicado como 16.567 bp, en la figura), con solución de continuidad (circular). Este ADN mitocondrial contiene la información codificada para la síntesis de 13 proteínas de la «cadena respiratoria», junto con los ARN ribosómicos y los ARN de transferencia para la traducción de todos los codones (tripletes de nucleótidos que codifican la adición de un aminoácido a la cadena proteica que se está sintetizando) No obstante, las mitocondrias también contienen proteínas codificadas por el ADN del núcleo celular. Así pues, las mitocondrias necesitan de otras estructuras celulares, al tiempo que son imprescindibles para la viabilidad de las propias células (endosimbiosis).
Las mitocondrias son orgánulos semiautónomos que mantienen una relación endosimbiótica con el «hospedador» (la propia célula). No en vano, estos orgánulos tienen su propio ADN, de un tamaño muy variable según las especies. El ADN mitocondrial humano tiene 16.569 pares de bases (indicado como 16.567 bp, en la figura), con solución de continuidad (circular). Este ADN mitocondrial contiene la información codificada para la síntesis de 13 proteínas de la «cadena respiratoria», junto con los ARN ribosómicos y los ARN de transferencia para la traducción de todos los codones (tripletes de nucleótidos que codifican la adición de un aminoácido a la cadena proteica que se está sintetizando) No obstante, las mitocondrias también contienen proteínas codificadas por el ADN del núcleo celular. Así pues, las mitocondrias necesitan de otras estructuras celulares, al tiempo que son imprescindibles para la viabilidad de las propias células (endosimbiosis).
¿Cómo surgió esa relación endosimbiótica?
Un organismo (procariota), capaz de realizar la fosforilación oxidativa («cadena respiratoria») fue engullido por una célula (eucariota).
Hay aspectos para justificar como plausible el modelo anterior de endosimbiosis, tales como la presencia en la mitocondria de una doble membrana, ADN circular; así como la existencia de «maquinarias» de transcripción y traducción autónomas. Está muy asentada la teoría del origen bacteriano de las mitocondrias. Hay razones para ello. El genoma bacteriano más parecido al de las mitocondrias es el de la bacteria Rickettsia prowazekii, causante del tifus transmitido por piojos (enfermedad de Brill-Zinsser). El genoma de Rickettsia porwazekii tiene 1 millón de pares de bases organizados en 834 genes que codifican proteínas. Hasta donde hoy se sabe, todas las mitocondrias proceden de un ancestro de Rickettsia prowazekii y son resultado de un «único» proceso de endosimbiosis.
¿Qué hace pensar que solo se produjo un único evento endosimbiótico?
La evidencia procede del estudio del genoma mitocondrial más parecido al bacteriano. Se trata del genoma del protozoo Reclinomomas americana. Su genoma se estructura en 97 genes; 62 de ellos codifican proteínas que representan la totalidad de los genes hallados en todos los genomas mitocondriales secuenciados. Aun así, este genoma representa menos del 2% de todos los genes que codifican proteínas de la bacteria Escherichia coli
No es extraño que una célula eucariota engulla a otra célula procariota. Bajo determinadas circunstancias, este proceso de depredación se adaptó a una simbiosis (endosimbiosis) que se ha consolidado evolutivamente.
Zaragoza, a 16 de julio de 2018
Dr. José Manuel López Tricas
Farmacéutico especialista Farmacia Hospitalaria
Farmacia Las Fuentes
Florentino Ballesteros, 11-13
50002 Zaragoza






























{kind=link}
{kind=link}