Es bien sabido el problema de la resistencia a los antibióticos. Incluidos los prescritos como último recurso en las infecciones más graves, los carbapenems, tienen su talón de Aquiles. Se trata de las enzimas metalo-beta-lactamasa New Dehli, y la metalo-beta-lactamasa codificada por el integrón Verona (en función de las dos ciudades donde se aislaron cepas bacterianas resistentes).
Un grupo de investigación de la universidad McMaster (Hamilton, Ontario, Canadá), dirigido por C.D. Wrigth puede haber hallado la estrategia para soslayar esta resistencia.
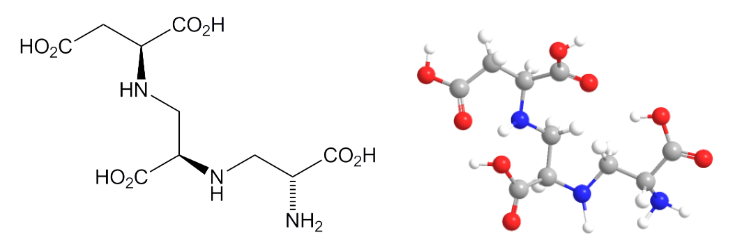
El moho (hongo) Aspergillus versicolor, prolífico en el suelo canadiense, sintetiza inusuales combinaciones de aminoácidos, incluido el ácido tetrahidrocarboxílico aspergilomarasmina-A. Dos de sus grupos ácidos provienen del ácido aspártico, otros dos de dos moléculas del aminoácido alanina. Una variante, aspergilomarasmina-B contiene el aminoácido glicina (glicocola) en lugar de una de las alaninas de la aspergilomarasmina-A.
Las enzimas metalo-beta-lactamasas precisan la presencia de zinc para su resistencia frente a los carbapenems. El grupo de investigación canadiense descubrió que la aspergilomarasmina-A puede eliminar el zinc sin dar lugar a efectos tóxicos. Así pues, la combinación de carbapenems y aspergilomarasmina-A permitió que los antibióticos siguieran siendo efectivos frente a bacterias anteriormente resistentes.
Zaragoza a 11 de febrero de 2019
Dr. José Manuel López Tricas
Farmacéutico especialista Farmacia Hospitalaria
Farmacia Las Fuentes
Florentino Ballesteros, 11-13
50002 Zaragoza























{kind=link}