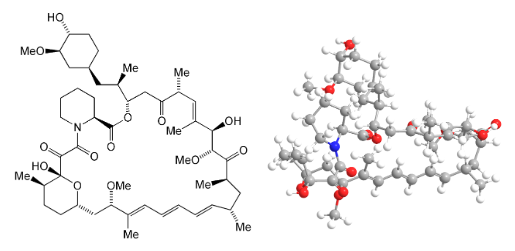
Los macrólidos, compuestos con estructura de lactonas cíclicas y con sustituyentes sacáridos reducidos, son productos de origen natural aislados a partir de bacterias. Algunos son eficaces antibióticos, como la eritromicina y la azitromicina.
Otro macrólido, la rapamicina, tiene historia larga y convulsa.
En el año 1964, Georges Nógrády, microbiólogo de la universidad de Montreal, Canadá, formó parte de una expedición científica a la Isla de Pascua, en el sur del océano Pacífico. Su misión era estudiar porqué los nativos no contraían la infección por tétanos, a pesar de las favorables condiciones edafológicas y climatológicas para el crecimiento de la bacteria, Clostridium tetanii. Para ello recopilaron muestras del terreno.
Tras examinar más de 60 muestras del suelo solo hallaron una única espora de la bacteria. Georges Nógrády envió las muestras recolectadas a Ayerst Pharmaceuticals, en Montreal. Siguiendo años de esmerado trabajo los investigadores hallaron que una bacteria producía un compuesto con actividad fungicida al que denominaron rapamicina, rebautizada más tarde como sirolimus. La primera denominación, rapamicina, hacía referencia a Rapa Nui, el topónimo de la Isla de Pascua en la lengua de los nativos.
Sirolimus (Rapamicina) mostró ser un eficaz inmunosupresor.
Sin embargo, en el año 1982 Ayerst Pharmaceuticals sufrió una reestructuración y el proyecto de investigación de la entonces todavía denominada rapamicina se vió interrumpido. Tras su integración en el laboratorio norteamericano Wyeth, formándose la división Wyeth-Ayerst, los proyectos en curso se reanudaron.
Fue necesario esperar hasta el año 1999, para que sirolimus (rapamicina) recibiera la autorización por la Food and Drug Administration estadounidense como medicamento inmunosupresor. Una modificación química de la molécula original se aprobó para el tratamiento del cáncer renal en el año 2007.
Dos años después, 2009, investigadores de Jackson Laboratories, en Main, Estados Unidos, descubrieron que sirolimus (rapamicina) prolongaba la supervivencia de los ratones, una especie de «elixir de juventud».
Hoy día sirolimus (rapamicina) – ahora ya propiedad de la multinacional Pfizer – forma parte del armamentaria para la prevención del rechazo tras el trasplante de órganos.
Zaragoza, a 8 de marzo de 2019
Dr. José Manuel López Tricas
Farmacéutico especialista Farmacia Hospitalaria
Farmacia Las Fuentes
Florentino Ballesteros, 11-13
50002 Zaragoza


























{kind=link}