
Es una genopatía recesiva asociada al cromosoma X, que solo afecta a varones, caracterizada por anormalidades conductuales y neurológicas. La alteración bioquímica que subyace en el síndrome es una acumulación de ácido úrico que da lugar a precipitados de uratos alcalinos, sobre todo urato sódico, en las articulaciones y túbulos renales, que se traduce clínicamente en artritis gotosa y cálculos renales respectivamente.
Las alteraciones neurológicas del «Síndrome de Lesch Nyhan» incluyen distonías, movimientos coreicos y balismo. Este cuadro lleva asociado conductas de autolesión que limitan la autonomía de las personas afectadas.
La incidencia estimada es de 1 caso cada 380.000 personas.
El síndrome fue descrito por Michael Lesch y William Nyhan en el año 1964 [Lesch M., Nyhan WL. A familial disorder of uric acid’ metabolism and central nervous system function. Am J. Med., 1964; 36(4): 561-70]. Edwin Seegmiller desentrañó la causa metabólica del síndrome [Seegmiller JE., Rosenbloom FM. Enzyme defect associated with a sex-linked human neurological disorder and excessive purine synthesis. Science 1967; 155(770):1682-4].
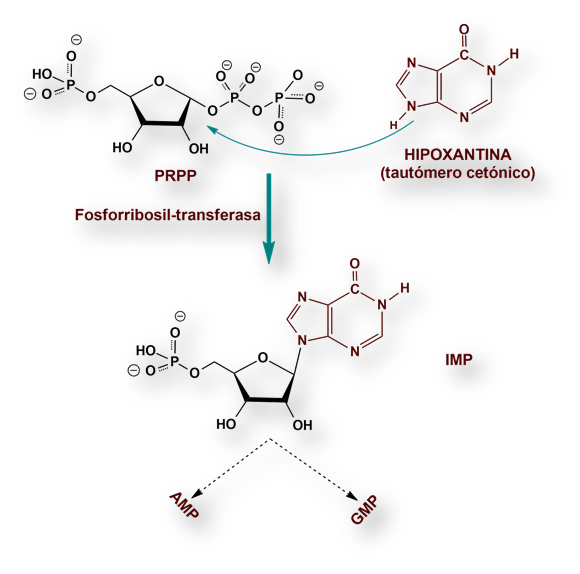 El «Síndrome de Lesch Nyhan» se debe a una mutación del gen HPRT1 que contiene la información para la síntesis de la enzima hipoxantina-fosforribosiltransferasa-1. Esta enzima cataliza la reacción entre la hipoxantina y el PRPP –Fosforribosilpirofosfato– para formar IMP (inosinato); y posteriormente AMP o GMP. De modo muy resumido, he aquí la síntesis «de novo» de las purinas (ver figura adjunta).
El «Síndrome de Lesch Nyhan» se debe a una mutación del gen HPRT1 que contiene la información para la síntesis de la enzima hipoxantina-fosforribosiltransferasa-1. Esta enzima cataliza la reacción entre la hipoxantina y el PRPP –Fosforribosilpirofosfato– para formar IMP (inosinato); y posteriormente AMP o GMP. De modo muy resumido, he aquí la síntesis «de novo» de las purinas (ver figura adjunta).
La mutación que causa el síndrome es un acortamiento (déficit parcial) o ausencia (déficit total) del gen. En caso de acortamiento, se sintetiza una versión defectuosa de la enzima que mantiene una actividad residual. Se produce un cuadro clínico con ausencia de uno de los síntomas más terribles del síndrome, la tendencia a la autolesión.
En ausencia del gen, los nucleótidos de purina no se pueden reciclar. El resultado es la metabolización de los nucleótidos, vía xantina, hasta uratos (sobre todo urato de sodio) [Ver bajo el epígrafe «Degradación de las purinas hasta urato», más adelante en este mismo texto]. Por razones ignoradas, un déficit de esta enzima se asocia con bajos niveles de dopamina en el tejido cerebral. La dopamina es un neurotransmisor fundamental para el control de los movimientos y la conducta. Sin embargo, no se ha desentrañado el mecanismo por el que una baja concentración de dopamina da lugar a los graves y destructivos síntomas del «Síndrome de Lesch Nyhan».
El gen HPRT1 se localiza en el cromosoma X. Los padres no pueden transmitir este rasgo a sus hijos. Solo las mujeres portadoras de un alelo (una de las copias del gen) mutado actúan como portadoras. En la descendencia de una pareja formada por un varón sano y una mujer portadora, la mitad de los hijos varones serán sanos, y la otra mitad padecerán el síndrome. Dicho de otra manera, cada hijo varón tendrá una probabilidad de estar sano del 50%, las mismas que de sufrir el síndrome La mitad de las mujeres nacerán sanas, y la otra mitad serán portadoras asintomáticas. O, dicho de otra manera, cada una de las dos situaciones tendrá una probabilidad de suceder del 50%.
Las características más dramáticas del «Síndrome de Lesch Nyhan» se manifiestan en la primera infancia, a partir del 2º o 3er año de vida, con conductas autodestructivas. Los niños comienzan a morderse los dedos o los labios hasta causarse graves lesiones, incluso amputaciones. Conforme crecen la conducta agresiva se expresa también hacia los demás niños. Pronto se hace evidente retraso mental y espasmos. Así mismo, aparecen cálculos renales y síntomas de gota (artritis gotosa).
La alteración bioquímica que subyace en esta genopatía es la total ausencia de actividad enzimática hipoxantina-fosforribosiltransferasa. La primera consecuencia es la acumulación de PRPP (Fosforribosil-pirofosfato), un incremento muy notable de la biosíntesis de purinas por la vía «de novo» y una desmesurada producción de uratos, sobre todo urato de sodio, de baja solubilidad, que termina precipitando en articulaciones y túbulos renales.
DEGRADACIÓN DE LAS PURINAS A URATO
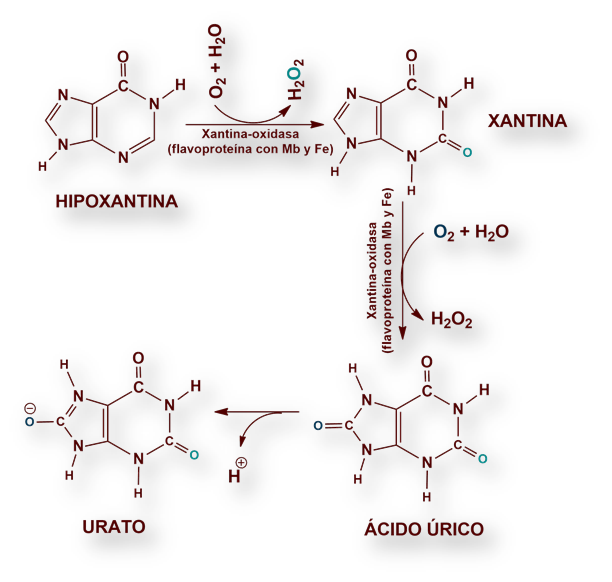 Algunas bases derivadas de la purina (adenina, guanina) se reutilizan para formar nucleótidos; otras se degradan hasta hipoxantina, mediante desaminación y ruptura hidrolítica del enlace glucosídico.
Algunas bases derivadas de la purina (adenina, guanina) se reutilizan para formar nucleótidos; otras se degradan hasta hipoxantina, mediante desaminación y ruptura hidrolítica del enlace glucosídico.
La enzima xantina-oxidasa (de la que existen versiones recombinantes como futuros medicamentos contra la gota refractaria – véase informe sobre «Tratamiento de la gota»), es una flavoproteína (FAD, como cofactor) que contiene molibdeno y hierro como grupos prostéticos.
La actividad xantina-oxidasa cataliza la conversión de hipoxantina a xantina; y ésta a ácido úrico. El O2 que actúa como oxidante en ambas reacciones, se convierte den H2O2, que se descompone posteriormente hasta H2O Y O2 por acción enzimática de la catalasa.
En los humanos el ácido úrico (en forma de uratos solubles) es el producto final de la degradación de las purinas. Se excreta en la orina. A concentraciones elevadas, el urato de sodio precipita en articulaciones y túbulos renales, dando lugar a los clásicos tofos de la artritis gotosa y cálculos renales.

 En los humanos la cantidad de urato (sobre todo urato de sodio) en el suero se halla muy próximo al límite de su solubilidad. No así en prosimios tales como lémures, en los que la concentración usual es 10 veces más baja que en los humanos. La filogenia dio lugar a un incremento de la concentración de urato hasta límites cercanos a su insolubilidad. ¿Qué ventaja evolutiva justifica el incremento de la concentración sérica de urato? El urato es un antioxidante metabólico tan eficaz como la vitamina C (ácido ascórbico). La estrategia para contrarrestar los oxidantes que se generan durante el metabolismo tiene su trasunto en mayor longevidad y protección contra las mutaciones que dan lugar a deriva cancerosa.
En los humanos la cantidad de urato (sobre todo urato de sodio) en el suero se halla muy próximo al límite de su solubilidad. No así en prosimios tales como lémures, en los que la concentración usual es 10 veces más baja que en los humanos. La filogenia dio lugar a un incremento de la concentración de urato hasta límites cercanos a su insolubilidad. ¿Qué ventaja evolutiva justifica el incremento de la concentración sérica de urato? El urato es un antioxidante metabólico tan eficaz como la vitamina C (ácido ascórbico). La estrategia para contrarrestar los oxidantes que se generan durante el metabolismo tiene su trasunto en mayor longevidad y protección contra las mutaciones que dan lugar a deriva cancerosa.
Zaragoza, a 23 de julio de 2018
Dr. José Manuel López Tricas
Farmacéutico especialista Farmacia Hospitalaria
Farmacia Las Fuentes
Florentino Ballesteros, 11-13
50002 Zaragoza