
RECEPTORES GABA
El ácido γ-aminobutírico (referido de manera usual por su acrónimo GABA, de Gamma Amino Butyric Acid) es un neurotransmisor ubicuo en el cerebro y médula espinal. Actúa como neurotransmisor inhibidor.
Su presencia en otros tejidos distintos del Sistema Nervioso Central es testimonial. Dentro del cerebro, su concentración varía de 2 a 5mcmol/g en la sustancia gris, siendo máxima (10mcmol/g) en la región nigroestriada. [La vía nigroestriada es una de las rutas cerebrales de transmisión dopaminérgica. Los somas neuronales se hallan en la sustancia nigra, y los axones se proyectan en el cuerpo estriado, una «estación de paso» de los impulsos nerviosos hacia los ganglios basales. La vía nigroestriada es primordial en el control motor. Las alteraciones de las vías dopaminérgicas nigroestriadas explican las alteraciones bioquímicas de varias enfermedades neurodegenerativas, siendo las más notables la enfermedad de parkinson y la enfermedad de Huntington; así como los efectos extrapiramidales de los antagonistas dopaminérgicos, tales como los fármacos antipsicóticos clásicos (neurolépticos y butirofenonas).
 GABA se sintetiza por descarboxilación enzimática del ácido glutámico (un aminoácido). La presencia de la enzima se usa para delimitar las vías de transmisión gabaérgica. Tras la liberación del neurotransmisor (GABA) al espacio sináptico, las moléculas son recapturadas por las neuronas presinápticas y astrocitos adyacentes. [Los astrocitos – células con prolongaciones que les hacen parecer un astro – de ahí su nombre -, junto con las células de Schwann y los oligodendrocitos, constituyen las denominadas genéricamente células de glía].
GABA se sintetiza por descarboxilación enzimática del ácido glutámico (un aminoácido). La presencia de la enzima se usa para delimitar las vías de transmisión gabaérgica. Tras la liberación del neurotransmisor (GABA) al espacio sináptico, las moléculas son recapturadas por las neuronas presinápticas y astrocitos adyacentes. [Los astrocitos – células con prolongaciones que les hacen parecer un astro – de ahí su nombre -, junto con las células de Schwann y los oligodendrocitos, constituyen las denominadas genéricamente células de glía].
Algunas sustancias inhiben este mecanismo de recaptación. Entre ellas se hallan: guvacina, ácido nipecótico y Tiagabina, este último un medicamento antiepiléptico


El GABA experimenta transaminación enzimática: el grupo amino se transfiere al α-oxoglutarato obteniéndose glutamato y hemialdehído succínico, oxidado finalmente hasta ácido succínico. La reacción es catalizada por la enzima GABA-transaminasa, localizada fundamentalmente en los astrocitos. Otro importante medicamento antiepiléptico, Vigabatrina, inhibe esta enzima (GABA-transaminasa).

 El GABA es un neurotransmisor inhibidor en múltiples vías del Sistema Nervioso Central. Aproximadamente el 20% de todas las neuronas del sistema nervioso tienen el GABA como neurotransmisor. Este hecho explica la importancia de la inhibición para el correcto funcionamiento del sistema nervioso. Una confirmación de este supuesto procede del efecto convulsionante de una sustancia inhibidora de la actividad gabaérgica cerebral, la bicuculina.
El GABA es un neurotransmisor inhibidor en múltiples vías del Sistema Nervioso Central. Aproximadamente el 20% de todas las neuronas del sistema nervioso tienen el GABA como neurotransmisor. Este hecho explica la importancia de la inhibición para el correcto funcionamiento del sistema nervioso. Una confirmación de este supuesto procede del efecto convulsionante de una sustancia inhibidora de la actividad gabaérgica cerebral, la bicuculina.
FARMACOLOGÍA DE LOS RECEPTORES DEL GABA
Se han identificado dos tipos de receptores para el ácido γ-aminobutírico: GABAA y GABAB. Un tercer tipo de receptor (GABAC) se considera hoy día un subtipo de receptor GABAA [Ver: Olsen, R.W., Sieghart W. International Union of Pharmacology. LXX. Subtypes of γ-aminobutyric acidA receptors: classification on basis of subunit composition, pharmacology, and function. Update. Pharmacol. Rev. 2008; 60: 243-260].
Receptores GABAA.-
 Los receptores GABAA pertenecen a la misma familia que los receptores para otros neurotransmisores, tales como glicina (véase más adelante, bajo el epígrafe Glicina, en este mismo texto), ácido nicotínico y serotonina (5HT3) [Barnard E.A. The molecular architecture of GABAA and Glycine neurotransmission. Handbook of Experimental Pharmacology. 2000. Springer-Verlag, Berlin; páginas 79-100].
Los receptores GABAA pertenecen a la misma familia que los receptores para otros neurotransmisores, tales como glicina (véase más adelante, bajo el epígrafe Glicina, en este mismo texto), ácido nicotínico y serotonina (5HT3) [Barnard E.A. The molecular architecture of GABAA and Glycine neurotransmission. Handbook of Experimental Pharmacology. 2000. Springer-Verlag, Berlin; páginas 79-100].
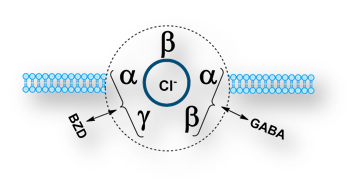
Los receptores GABAA son pentámeros. Se han clonado varias subunidades. Visualizado desde el medio extracelular, el pentámero GABAA tiene una estructura cuaternaria «α-β-α-β-γ» que conforma un poro central (canal para los aniones Cl–).
El ligando fisiológico (GABA) se une en la interfaz α/β. Las benzodiacepinas (BZD) y compuestos relacionados, se engarzan en la interfaz α/γ. Las diferencias estructurales de las subunidades α y γ de los subtipos de receptores determinan las diferencias se sensibilidad a las distintas benzodiacepinas, hecho que podría explicar las respuestas idiosincrásicas de estos medicamentos.
Los receptores GABAA se localizan en las membranas de las neuronas post-sinápticas. Su activación, tras la unión de su ligando fisiológico (o sosias farmacológicas), aumenta la permeabilidad a los aniones Cl¯, hiperpolarizándose la membrana, con la consiguiente disminución de la excitabilidad.
 Los receptores del GABA se localizan tanto en sinapsis como en ubicaciones extrasinápticas. Estas últimas dan lugar a una inhibición «a distancia», una neuromodulación alejada del punto donde se segregó la molécula. Los receptores extrasinápticos del GABAA tienen una estructura cuaternaria «α4/α5/α6/δ». Estos receptores extrasinápticos son muy sensibles a los anestésicos y al etanol. Además tienen una mayor afinidad por el GABAA y muestran menos desensibilización. Gaboxadol (THIP), un agonista parcial del receptor GABAA, muestra afinidad por la subunidad δ.
Los receptores del GABA se localizan tanto en sinapsis como en ubicaciones extrasinápticas. Estas últimas dan lugar a una inhibición «a distancia», una neuromodulación alejada del punto donde se segregó la molécula. Los receptores extrasinápticos del GABAA tienen una estructura cuaternaria «α4/α5/α6/δ». Estos receptores extrasinápticos son muy sensibles a los anestésicos y al etanol. Además tienen una mayor afinidad por el GABAA y muestran menos desensibilización. Gaboxadol (THIP), un agonista parcial del receptor GABAA, muestra afinidad por la subunidad δ.
Receptores GABAB.-
 Los receptores GABAB se localizan pre- y post-sinápticamente [Bettler B., et al. Molecular structure and function of GABAB receptors. Physiol. Rev. 2004; 84: 835-67].
Los receptores GABAB se localizan pre- y post-sinápticamente [Bettler B., et al. Molecular structure and function of GABAB receptors. Physiol. Rev. 2004; 84: 835-67].
Los receptores GABAB pertenecen a la clase de proteínas C acopladas con proteínas G que regulan la permeabilidad transmembrana a los cationes Ca 2+. Consecuencia: disminuye la excitabilidad post-sináptica, con subsiguiente inhibición de la actividad enzimática adenilato-ciclasa. Esta enzima (adenilato-ciclasa) cataliza la hidrólisis de ATP ® AMPC (la principal fuente de energía metabólica en forma de cationes fosfato).
El receptor GABAB está formado por dos dímeros: GABAB1 y GABAB2. Las colas de estos dos dímeros atraviesan la bicapa lipídica de la membrana. Cuando el GABA (ligando fisiológico) se engarza con el dominio extracelular de la subunidad GABAB1 se produce un cambio conformacional en la subunidad GABAB2 que activa una vía de señalización celular (vía proteínas G) [Kubo & Tateyana. Current Opinion in Neurology. 2005; 15: 289-295].
Fármacos que interactúan en el receptor GABAA.-
Los receptores para el GABAA remedan a los receptores NMDA (N-Metil-D-Aspartato) en que los fármacos interactúan con el receptor en un lugar distinto del centro activo. En base a esta circunstancia se subdividen en varios subtipos. [Johnston G.A.R. GABAA-receptor phamacology. Pharmacol. Ther. 1996; 69: 173-198].
Los receptores GABAA son la diana farmacológica de varios y trascendentes fármacos de acción central: benzodiacepinas y análogos, barbitúricos, neuroesteroides y varios anestésicos generales.
 Muscinol, extraído de un hongo alucinógeno, es un potente agonista del receptor GABAA. Un derivado semisintético del Muscinol, denominado Gaboxadol, es un agonista parcial que llegó a comercializarse durante algún tiempo como hipnótico. Su papel en terapéutica fue breve dado que no representaba ninguna mejora en relación a las benzodiacepinas.
Muscinol, extraído de un hongo alucinógeno, es un potente agonista del receptor GABAA. Un derivado semisintético del Muscinol, denominado Gaboxadol, es un agonista parcial que llegó a comercializarse durante algún tiempo como hipnótico. Su papel en terapéutica fue breve dado que no representaba ninguna mejora en relación a las benzodiacepinas.
Bicuculina es un compuesto de origen natural aislado en 1932, con actividad antagonista sobre el receptor GABAA. Se utiliza como inductor de convulsiones en el screening farmacológico de potenciales medicamentos antiepilépticos.
Las benzodiacepinas de engarzan a un lugar accesorio del receptor GABAA definido como «receptor benzodiacepínico» (interactúa con el dímero α/γ del pentámero que constituye el receptor GABAA). La unión de la molécula de benzodiacepina (y compuestos relacionados) facilita la unión de los agonistas con el receptor. El resultado final es un aumento del efecto inhibidor derivado de la activación de los receptores GABAA.
Los «moduladores del receptor GABAA» se unen al receptor en un lugar diferente a donde lo hacen las benzodiacepinas y análogos. En el grupo de los «moduladores del canal» central del receptor GABAA se incluyen los barbitúricos, algunos anestésicos generales y los neuroesteroides (metabolitos de los andrógenos y de la progesterona).
 Un neuroesteroide sintético, alfaxalona, es un anestésico general.
Un neuroesteroide sintético, alfaxalona, es un anestésico general.
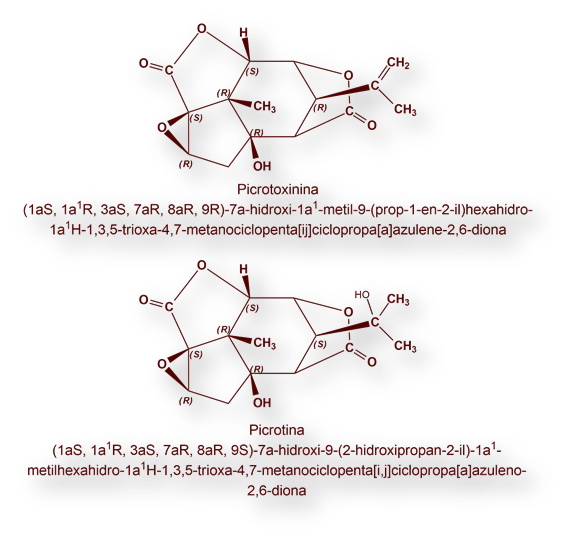 Picrotoxina (mezcla equimolar de Picrotoxinina y Picrotina) es una sustancia convulsionante que bloquea el canal para el anión Cl– asociado al receptor GABAA. De esta guisa, se bloquea el efecto inhibidor postsináptico de las neuronas gabaérgicas.
Picrotoxina (mezcla equimolar de Picrotoxinina y Picrotina) es una sustancia convulsionante que bloquea el canal para el anión Cl– asociado al receptor GABAA. De esta guisa, se bloquea el efecto inhibidor postsináptico de las neuronas gabaérgicas.
Fármacos que interactúan en el receptor GABAB.-
 Cuando se descubrió la actividad inhibidora de la función cerebral del GABA, se iniciaron varias líneas de investigación dirigidas a sintetizar moléculas que remedaran la actividad del GABA como potenciales fármacos antiepilépticos. Uno de los problemas era sintetizar moléculas más liposolubles que el GABA, con mejor biodisponibilidad en el tejido cerebral. Fruto de esta investigación fue la síntesis del Baclofeno, introducido en terapéutica en el año 1972. A diferencia del GABA, la acción del Baclofeno no es anulada por la Bicuculina. Este hallazgo fue determinante para la aceptación de un segundo tipo de receptor gabaérgico: GABAB. Baclofeno se prescribe para el tratamiento de la espasticidad en los desórdenes motores.
Cuando se descubrió la actividad inhibidora de la función cerebral del GABA, se iniciaron varias líneas de investigación dirigidas a sintetizar moléculas que remedaran la actividad del GABA como potenciales fármacos antiepilépticos. Uno de los problemas era sintetizar moléculas más liposolubles que el GABA, con mejor biodisponibilidad en el tejido cerebral. Fruto de esta investigación fue la síntesis del Baclofeno, introducido en terapéutica en el año 1972. A diferencia del GABA, la acción del Baclofeno no es anulada por la Bicuculina. Este hallazgo fue determinante para la aceptación de un segundo tipo de receptor gabaérgico: GABAB. Baclofeno se prescribe para el tratamiento de la espasticidad en los desórdenes motores.
Los antagonistas competitivos del receptor del GABAB incluyen diversas sustancias sin utilidad clínica, tales como 2-hidroxisaclofen, CGP35348. Estos compuestos (antagonistas del receptor GABAB) ejercen mínimos efectos sobre la función cerebral, a diferencia de los antagonistas del receptor GABAA.
γ-HIDROXI-BUTIRATO.-
 El γ-hidroxibutirato está presente en el tejido cerebral. Es un precursor en la biosíntesis del γ-aminobutirato (forma ionizada del ácido γ-aminobutírico, o GABA) [Wrong C.G.T., et al. From street to brain: neurobiology of the recreational drug γ-hidroxibutyric acid. Trends Pharmacol Sci. 2004; 25: 29-34]. A partir de su síntesis en la década de 1960, esta sustancia se usa ilegalmente en ambientes de culturismo (por su supuesta capacidad para estimular la secreción de somatotropina u hormona de crecimiento), y en ambientes «festivos», por su acción euforizante. Es un agonista débil del receptor GABAB [Wu Y., et al. γ-Hidroxibutyric acid (GHB) and γ-hidroxibutyric acid B receptor binding site are distinctive. Neuropharmacology 2004; 47: 1146-1156]. Algunos estudios muestran que activa los circuitos neuronales de recompensa, que involucra vías neuronales dopaminérgicas en varias estructuras cerebrales (amígdala, núcleo accumbens, área de Tsai y cerebelo); así como neuronas oxitocínicas en la hipófisis o glándula pituitaria.
El γ-hidroxibutirato está presente en el tejido cerebral. Es un precursor en la biosíntesis del γ-aminobutirato (forma ionizada del ácido γ-aminobutírico, o GABA) [Wrong C.G.T., et al. From street to brain: neurobiology of the recreational drug γ-hidroxibutyric acid. Trends Pharmacol Sci. 2004; 25: 29-34]. A partir de su síntesis en la década de 1960, esta sustancia se usa ilegalmente en ambientes de culturismo (por su supuesta capacidad para estimular la secreción de somatotropina u hormona de crecimiento), y en ambientes «festivos», por su acción euforizante. Es un agonista débil del receptor GABAB [Wu Y., et al. γ-Hidroxibutyric acid (GHB) and γ-hidroxibutyric acid B receptor binding site are distinctive. Neuropharmacology 2004; 47: 1146-1156]. Algunos estudios muestran que activa los circuitos neuronales de recompensa, que involucra vías neuronales dopaminérgicas en varias estructuras cerebrales (amígdala, núcleo accumbens, área de Tsai y cerebelo); así como neuronas oxitocínicas en la hipófisis o glándula pituitaria.
GLICINA.-
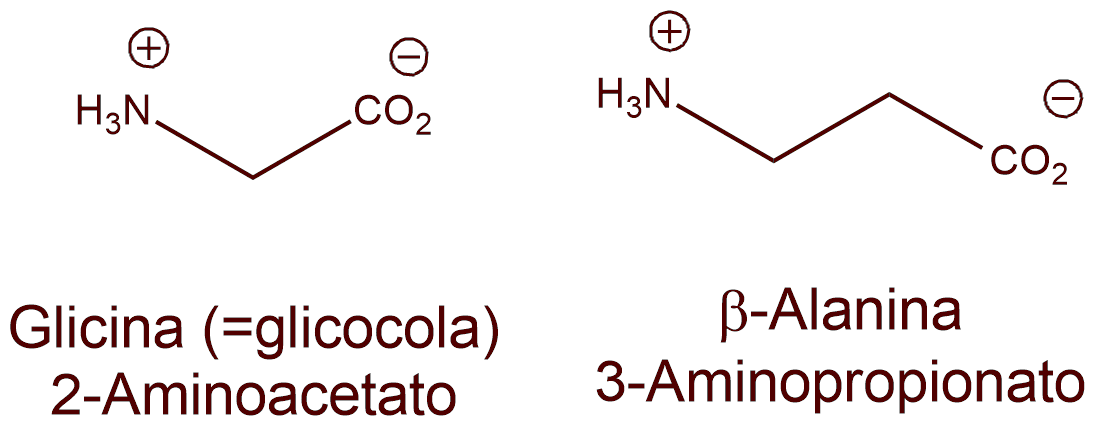 La glicina (o glicocola) se halla presente, a una concentración de 5mcmol/g, en la materia gris de la médula espinal.
La glicina (o glicocola) se halla presente, a una concentración de 5mcmol/g, en la materia gris de la médula espinal.
Cuando se aplica mediante iontoforesis a las neuronas motoras e interneuronas medulares, se hiperpolarizan, de un modo indistinguible, a una respuesta sináptica inhibidora.
La estricnina, un tóxico de origen vegetal, actúa en la médula espinal. Anula la actividad inhibidora de la glicina en las interneuronas y neuronas motoras medulares, dando lugar a cuadros convulsivos.
β-ALANINA.-
La β-alanina tiene una distribución similar a la glicina; y, al igual que ésta, ejerce acción inhibidora de la transmisión nerviosa. A diferencia de la glicina, la actividad inhibidora de la β-alanina no se contrarresta por la estricnina.
La toxina del tétanos remeda a la toxina botulínica en que actúa de modo selectivo previniendo la liberación de glicina de las interneuronas de la médula espinal. Como consecuencia de produce hiperexcitabilidad refleja y violentos espasmos, entre ellos el mandibular (patognomónico de la toxemia tetánica).
´La glicina (=glicocola) se elimina del espacio extracelular mediante dos transportadores específicos: GlyT1 y GlyT2. [Eulenburg V., et al. Glycine transporters: essential regulators of neurotransmission. Trends. Biochem. Sci. 2005; 30: 325-333].
GlyT1 se localiza en los astrocitos en todo el sistema nervioso central.
GlyT2 se ubica en neuronas glicinérgicas de la médula espinal, tronco cerebral y cerebelo.
La glicina no solo actúa como neurotransmisor inhibidor, sino como coadyuvante agonista del glutamato en los receptores NMDA. La inhibición de la captación de glicina (mediado por el transportador GlyT1) da lugar a un incremento de la concentración de glicina extracelular en el tejido nervioso, potenciando la actividad agonista del glutamato sobre sus receptores (NMDA). La inhibición del transportador GlyT1 podría ser útil en el tratamiento de la esquizofrenia y algunos síndromes álgicos.
Zaragoza, a 16 de junio de 2018
Dr. José Manuel López Tricas
Farmacéutico especialista Farmacia Hospitalaria
Farmacia Las Fuentes
Florentino Ballesteros, 11-13
50002 Zaragoza