
Propafenona es un medicamento anti-arrítmico que se encuadra en la clase IC siguiendo la clasificación Vaughan Williams. El informe que se estructura en las páginas siguientes es una revisión de tipo farmacéutico sin aspiraciones a un análisis clínico. Este fármaco ha de ser prescrito por un médico especialista en Cardiología.
FARMACODINAMIA
Acciones electrofisiológicas de la Propafenona.-
Bloqueo de la corriente de Na+. Se logra así una disminución dosis-dependiente de la frecuencia de despolarizaciones (potenciales de acción), con enlentecimiento de la conducción en el haz His-Purkinje (acción dromotropa negativa). Su trasunto electrocardiográfico es una onda QRS de mayor amplitud. Este mecanismo de acción clasifica a la Propafenona dentro del grupo IC, según la clasificación de Vaughan Williams de los medicamentos anti-arrítmicos. [Se han propuesto otras clasificaciones, tales como Sicilian Gambit, pero no suponen mejoras a la clásica de Vaughan Williams, que si bien surgió con fines de investigación en farmacología, los Clínicos la utilizan de modo rutinario en terapéutica].
Propafenona no modifica el intervalo QT (repolarización ventricular).
Acciones hemodinámicas de la Propafenona.-
Tanto Propafenona como su metabolito activo (5-hidroxilado) tienen efecto inotropo negativo, esto es disminuyen la «fracción de eyección ventricular» (% de reducción del volumen ventricular durante la sístole). Si la «fracción de eyección» del paciente es normal o está moderadamente reducida (en no más de un 40%), el efecto inotropo negativo no es suficiente para causar insuficiencia cardíaca. Pero, por esta misma razón, Propafenona está contraindicado si la «fracción de eyección ventricular» es ≤30% del valor normal.
Efecto β-bloqueante de la Propafenona.-
Propafenona tiene un efecto β-bloqueante que oscila entre el 0,0125 y el 0,05 el del Propranolol (fármaco patrón de los β-bloqueantes). El efecto β-bloqueante de la Propafenona está asociado al enantiómero S. Este efecto β-bloqueante puede ser clínicamente significativo en los pacientes «metabolizadores lentos» del fármaco (esto es, aquellos que oxidan lentamente Propafenona a 5-hidroxi-propafenona).
Fármacos anti-arrítmicos de la clase I en la clasificación de Vaughan Williams.-
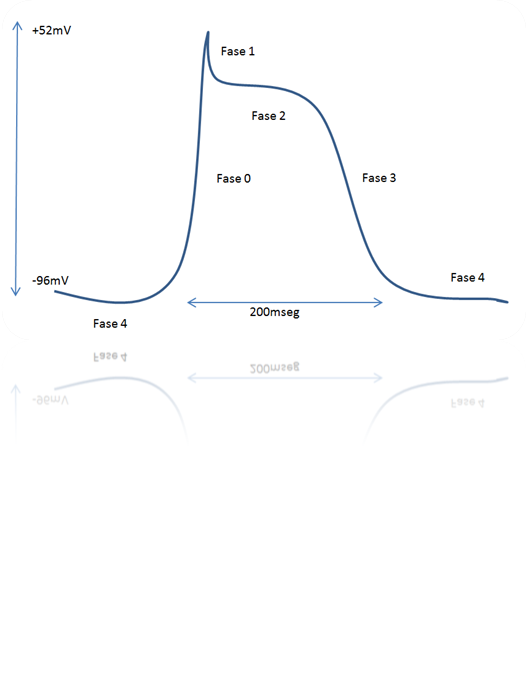 En estado de reposo, las células excitables tienen una diferencia de potencial trans-membrana con carga negativa neta en la cara interna de la membrana, y positiva en la cara externa. La diferencia de potencial está determinada por las distintas concentraciones iónicas a ambos lados de la membrana. El voltaje (diferencia de potencial) para cada ion se calcula mediante la ecuación de Walther Hermann Nernst:
En estado de reposo, las células excitables tienen una diferencia de potencial trans-membrana con carga negativa neta en la cara interna de la membrana, y positiva en la cara externa. La diferencia de potencial está determinada por las distintas concentraciones iónicas a ambos lados de la membrana. El voltaje (diferencia de potencial) para cada ion se calcula mediante la ecuación de Walther Hermann Nernst:
Voltaje = 61 x log (Concentración intracelular/Concentración extracelular)
[Walther Hermann Nernst fue galardonado con el Premio Nobel de Química en el año 1920, si bien no se le entregó hasta el año siguiente, 1921. Recuérdese que en el año 1920 la Fundación Alfred Nobel decidió que ninguno de los nominados cumplía las exigencias del testamento de Alfred Nobel. Los premios otorgados en 1920 fueron entregados el año siguiente].
La onda de despolarización (potencial de acción) altera rápida y transitoriamente la diferencia de potencial a ambos lados de la membrana. La diferencia de potencial pasa de -96mV a +52mV, restaurándose de nuevo la diferencia de potencial del estado de reposo al cabo de aproximadamente 200 mili-segundos.
De modo muy simplificado, las corrientes iónicas trans-membrana que acontecen secuencialmente durante el potencial de acción son las siguientes:
Fase 0: Na+ (out –> in)
Fase 1: K+ (in –> out)
Fase 2: Ca2+ (out –> in)
Fase 3: K+ (in –> out); Na+ (in –> out)
Fase 4: Na+ <—> K+ [intercambio iónico catalizado por la hidrólisis del ATP]
La función «potencial de acción vs tiempo» varía en las diferentes estirpes celulares excitables (nodo seno-auricular, nodo aurícula-ventrículo, haz de His-Purkinje y tejido ventricular). Las variaciones de la función «potencial de acción vs tiempo» (esto es, la forma de la curva) están condicionadas por la importancia de la corriente de cationes Ca2+ en cada localización.
Todos los medicamentos anti-arrítmicos clase I bloquean los canales de Na+. Se subdividen en tres grupos:
Los fármacos anti-arrítmicos clase I bloquean los canales de Na+ de modo similar a los anestésicos locales. De hecho, los anestésicos locales tienen propiedades anti-arrítmicas. Debido a que inhiben la propagación del potencial de acción en muchas células excitables en algunos textos antiguos de farmacología se hacía referencia a ellos como «estabilizadores de membrana», descripción hoy en desuso a favor de la que hace referencia de modo más preciso a su mecanismo de acción («bloqueantes de los canales de Na+»). La principal característica es disminuir la velocidad de despolarización durante la fase 0 del potencial de acción.
¿Por qué la clase I de la clasificación de Vaughan Williams se subdivide en tres subgrupos, IA, IB, IC? La razón es que los primeros medicamentos «bloqueantes de los canales de Na+» (Quinidina y Procainamida) tenían acciones electrofisiológicas que diferían de los más recientes. Todos los medicamentos anti-arrítmicos clase I bloquean la excitación miocárdica de alta frecuencia sin modificar la frecuencia fisiológica (el latido cardíaco normal). Los canales de Na+ existen tienen tres estados funcionales: cerrado, abierto y refractario. Ante la llegada de una onda de despolarización (potencial de acción) los canales cambian rápidamente del estado cerrado al abierto (activación). La despolarización mantenida, como sucede en un músculo isquémico, hace que el estado del canal de Na+ cambie del estado abierto al estado refractario (inactivación). La membrana ha de re-polarizarse de nuevo (reorganización de las cargas eléctricas a ambos lados de la membrana) para que el canal adquiera nuevamente el estado de cerrado y se halle en condiciones de abrirse con una nueva onda de despolarización.
Los fármacos anti-arrítmicos clase IA bloquean preferencialmente el canal de Na+ cuando se halla en estado abierto o refractario; y mucho menos en el estado cerrado. Esto establece una dependencia de uso (cuanto más activado se halle el canal mayor será el grado de bloqueo logrado con el medicamento). Por lo tanto, los fármacos anti-arrítmicos clase IA retrasan la re-polarización y prolongan la duración del potencial de acción.
Los fármacos anti-arrítmicos clase IB (Lidocaína) se asocian y disocian del canal rápidamente durante el latido normal. El fármaco se engarza al canal abierto durante la fase 0 del potencial de acción dejando bloqueado el canal durante el tiempo (milisegundos) que dura el potencial de acción. La disociación de las moléculas de fármaco dejará la membrana preparada para un nuevo potencial de acción, pero abortará cualquier latido prematuro. Así pues, aceleran la re-polarización y acortan la duración del potencial de acción.
Los fármacos anti-arrítmicos clase IC (Flecainida, Encainida, Propafenona) se asocian y disocian de los canales de Na+ mucho más lentamente alcanzando un grado de bloqueo que no varía sustancialmente durante el ciclo cardiaco. De este modo, los medicamentos de la clase IC inhiben de modo muy notorio la conducción del impulso cardíaco por el haz His-Purkinje. Ni aceleran ni retrasan la re-polarización y, consiguientemente el potencial de acción.
Adjuntamos la tabla siguiente a modo de resumen:
|
Clase I (Vaughan Williams) |
Repolarización |
Duración potencial acción |
Medicamentos |
|
|
IA |
Retrasan |
Alargan |
Procainamida, Disopiramida |
|
|
IB |
Aceleran |
Retrasan |
Lidocaína, Fenitoína |
|
|
IC |
No afectan |
No afectan |
Propafenona, Flecainida |
|
 Imagen típica de un electrocardiograma: el «pico» QRS corresponde con la despolarización (fase 0) del potencial de acción; y el segmento ST con la repolarización (fase 3).
Imagen típica de un electrocardiograma: el «pico» QRS corresponde con la despolarización (fase 0) del potencial de acción; y el segmento ST con la repolarización (fase 3).
FARMACOCINÉTICA
Absorción.-
Aproximadamente el 95% de la dosis oral de Propafenona contenida en el medicamento Rytmonorm® se absorbe, obteniéndose las concentraciones máximas en plasma (TMÁX) al cabo de entre 2 y 3 horas. Sin embargo, el fármaco experimenta un extenso «metabolismo de primer paso hepático» a dosis bajas. Así, la biodisponibilidad de dosis de entre 150 y 300mg varía del 5 al 12%; mientras es de entre un 40 y un 50% con dosis de 450mg. La función matemática «dosis vs CSS» es no-lineal. Por ejemplo, un aumento de la dosis por un factor de 3 (de 300 a 900mg) da lugar a un aumento de las concentraciones del estado de equilibrio (CSS) por un factor de 10.
Metabolismo.-
Propafenona se metaboliza casi completamente, recuperándose en orina menos del 1% de cada dosis administrada.
La Vida Plasmática Media (T1/2) es de 6 horas [rango: 2-10 horas].
Se han descrito hasta 11 metabolitos, aunque solo dos de ellos (5-hidroxi-propafenona y N-despropil-propafenona) son relevantes. El metabolismo oxidativo utiliza un sistema enzimático conocido como P450 dbl. Este sistema enzimático, responsable de la hidroxilación, está determinado fenotípicamente, estableciéndose dos grupos: «metabolizadores rápidos» y «metabolizadores lentos». Las características fenotípicas de la hidroxilación tienen trascendentes implicaciones tanto en la farmacocinética como en la incidencia de efectos adversos (véase más adelante).
El metabolito 5-hidroxi-propafenona contribuye de modo significativo a la acción anti-arrítmica de la Propafenona. La cinética de hidroxilación es saturable. El incremento de concentración de la Propafenona con la administración de dosis crecientes no se ajusta a una cinética lineal. Así pues, la T1/2 de la Propafenona en «metabolizadores rápidos» se halla en el rango 2 a 10 horas; mientras este rango varía entre 10 y 32 horas en el grupo de los «metabolizadores lentos».
Distribución.-
Unión a proteínas plasmáticas ≈ 80 – 90%.
Volumen Aparente de Distribución (VD) ≈ 1,1 – 3,6L/Kg.
Se observa una mayor amplitud de la onda QRS del electrocardiograma concordante con la captación del fármaco por el tejido miocárdico (vida media de captación ≈ 22 minutos).
No existe correlación entre las concentraciones plasmáticas de Propafenona y los efectos terapéuticos (anti-arrítmicos) y adversos.
DOSIFICACIÓN
La posología habitual es 150mg t.i.d. a 300mg t.i.d. (dosis máxima diaria: 900mg). Para el ajuste de la dosis se tiene más en cuenta la tolerancia al medicamento que las concentraciones plasmáticas. Otro factor a considerar es la extensión del intervalo QRS del electrocardiograma, que no debe incrementarse en más del 25% en relación al valor basal (antes de instaurar el tratamiento).
Para el tratamiento intravenoso: iniciar con una dosis de carga (Bolus) de 1,5 a 2 (mg/Kg); seguidos por una infusión intravenosa a razón de 0,007 a 0,014 (mg/Kg, minuto).
Hay que reducir la dosis en pacientes con insuficiencia renal y/o hepática.
Los comprimidos tienen sabor amargo y un ligero efecto anestésico. Han de tragarse enteros, sin chupar o masticar, junto con algo de líquido.
La Propafenona se considera contraindicada si se presentan alguna de las siguientes circunstancias:
- Fallo cardíaco congestivo no controlado.
- Disfunción ventricular izquierda («fracción de eyección» ≤ 30%).
- Disfunción del nodo atrial.
- Alteraciones de la conducción atrio-ventricular.
- Asma (la Propafenona tiene actividad β-bloqueante).
- Miastenia gravis (debido a la acción β-bloqueante de la Propafenona).
EFECTOS ADVERSOS
Son de tipo cardiovascular, gastrointestinal y sobre el sistema nervioso central.
La mayoría de los efectos adversos que no contraindiquen el uso de Propafenona se controlan disminuyendo la dosis prescrita. Es muy raro que lleguen a requerir la interrupción del tratamiento.
En situaciones de sobredosis, la administración de lactato sódico revierte los efectos electrofisiológicos y hemodinámicos de la Propafenona.
Los efectos adversos de tipo cardiovascular son más comunes en pacientes con enfermedad cardíaca avanzada y/o cuando se administran dosis elevadas. En estas circunstancias la incidencia es del 13 al 27%, según los estudios de fármaco-vigilancia.
Los dos efectos adversos de tipo cardíaco son: desencadenamiento de arritmias con riesgo de muerte súbita, y fallo cardíaco congestivo (sobre todo cuando la «fracción de eyección» ventricular está muy reducida). Por esta razón, el empleo de la Propafenona se considera inadecuado en estas situaciones.
Efectos adversos no-cardíacos de la Propafenona.-
Son de tipo neurológico (vértigo, disgeusia, visión borrosa, cefalea y parestesias); y gastrointestinales (náusea, vómito, estreñimiento, anorexia). Los «metabolizadores lentos de la debrisoquina» tienen una incidencia más elevada de efectos adversos neurológicos, relacionado con concentraciones plasmáticas más elevadas de Propafenona.
 Debrisoquina es un sustrato para el citocromo P450 isoenzima 2D6, un enzima polimórfico codificado por un gen mapeado en el cromosoma 22. Los pacientes homocigotos para este gen (los dos alelos mutados) son «metabolizadores lentos», expresando una actividad enzimática escasa o nula. La prevalencia del fenotipo «metabolizadores lentos» afecta al 5% aproximadamente de la población, de preferencia de raza blanca.
Debrisoquina es un sustrato para el citocromo P450 isoenzima 2D6, un enzima polimórfico codificado por un gen mapeado en el cromosoma 22. Los pacientes homocigotos para este gen (los dos alelos mutados) son «metabolizadores lentos», expresando una actividad enzimática escasa o nula. La prevalencia del fenotipo «metabolizadores lentos» afecta al 5% aproximadamente de la población, de preferencia de raza blanca.
Los «metabolizadores lentos» son incapaces de hidroxilar la debrisoquina sobre el átomo 5. Como la debrisoquina tiene actividad antihipertensiva, los «metabolizadores lentos» manifiestan una poderosa acción hipotensora cuando se administra debrisoquina. Este producto se ha usado para estudios fenotípicos de metabolización. No obstante los análisis basados en el ADN son más seguros y predecibles. Más información sobre polimorfismo metabólico de fármacos en el siguiente link: https://www.ncbi.nlm.nih.gov/pubmed/2689060?dopt=Abstract. Debrisoquina ya no se utiliza como medicamento anti-hipertensivo.
USO DE PROPAFENONA EN DISTINTOS TIPOS DE ARRITMIAS
- Taquicardia ventricular no mantenida
- Arritmia ventricular maligna
- Taquicardia ventricular mantenida (tratamiento a corto plazo)
- Cardioversión farmacológica de la fibrilación atrial
- Prevención de la fibrilación atrial recurrente
- Taquicardia atrio-ventricular de re-entrada
- Taquicardia supra-ventricular recurrente
Para información detallada de cada una de estas indicaciones, consultar con un médico especialista en Cardiología.
VALORACIÓN FARMACÉUTICA DE LA PROPAFENONA
La prescripción de Propafenona se ha de realizar teniendo en cuenta varios factores: (1º) tipo de arritmia y su prognosis; (2º) función ventricular izquierda (criterio «fracción de eyección ventricular»); (3º) alteraciones previas de la conducción cardíaca; y (4º) parámetros de función hepática y/o renal.
Los resultados del estudio clínico CAST (Cardiac Arrest Study Hamburg) han hecho reconsiderar los criterios de prescripción de los anti-arrítmicos clase I (recuérdese, Propafenona pertenece a la clase IC). Se considera que el tratamiento con Propafenona se debería restringir a pacientes con grave sintomatología SIN enfermedad cardíaca isquémica y CON suficiente «fracción de eyección» (> 30%). El objetivo del tratamiento busca más la abolición de los síntomas, y no tanto una mejora de la prognosis.
En pacientes con arritmias ventriculares, los medicamentos anti-arrítmicos tienen una eficacia limitada, además de ser pro-arrítmicos.
Los resultados de los ensayos clínicos ESVEM (Electrophysiological Studies Versus Electrocardiographic Monitoring) y CASH (Cardiac Arrest Study Hamburg) han revalorizado el uso de Sotalol como medicamento anti-arrítmico. [Sotalol tiene acciones de la clase II (β-bloqueante) y de la clase III (prolongación del potencial de acción, evidenciado por la extensión del intervalo PR del electrocardiograma].
Durante los últimos años Propafenona se ha prescrito cada vez con más frecuencia en las arritmias supra-ventriculares, bien como tratamiento de corta duración o de manera profiláctica.
Propafenona también se usa en pacientes con el síndrome de Wolf-Parkinson-White para prevenir la conducción anterógrada. [Síndrome de Wolf-Parkinson-White es una alteración congénita de la conducción cardíaca entre la aurícula y el ventrículo. Se evidencia electrocardiográficamente por una onda extra, designada por la letra griega δ).
La acción β-bloqueante de la Propafenona es muy escasa, excepto en los «metabolizadores lentos».
Zaragoza, a 15 de febrero de 2017
Dr. José Manuel López Tricas
Farmacéutico especialista Farmacia Hospitalaria
FARMACIA LAS FUENTES
ZARAGOZA