
La Agencia de Alimentos y Fármacos estadounidense (US FDA, de United States Food and Drug Administration) ha reconocido que el fabricante de la primera, y costosísima primera terapia génica, para tratar a niños con atrofia muscular espinal ocultó primero, y retrasó deliberadamente después, la publicación de información durante el proceso administrativo de aprobación de Zolgensma®.
Zolgensma® es nombre registrado de Onasemnogene abeparvovec xioi. También se designa: AAV9-CBA-SMN1-gene therapy Avexis (Adeno-associated-serotype-9-Chicken-Betha-Actin-Survival-Motor-Neuron-gene-therapy AveXis). [AveXis, es la división de la multinacional helvética Novartis AG, que desarrolló esta terapia génica].
La manipulación motivo de la controversia no tiene implicaciones clínicas, refiriéndose a la transcripción de los resultados de la experimentación en animales (ratones).
El tratamiento con Zolgensma® (una única perfusión) tiene un coste de 2,1 millones de dólares. El problema del coste es que crea un precedente ante próximos tratamientos para multitud de enfermedades raras. Este tratamiento fue aprobado en Estados Unidos en mayo de 2019.
Aun cuando el laboratorio ha reafirmado que los niños afectados de atrofia muscular espinal pueden recibir el tratamiento, tratándose de una terapia génica tan costosa y pionera, otros fabricantes que tienen en cartera terapias génicas para enfermedades raras pueden ver comprometido su prestigio y credibilidad.
La Food and Drug Administration ha otorgado un gran número de autorizaciones por vía de urgencia para tratamientos innovadores (casi siempre únicos) para enfermedades raras. Zolgensma® fue uno de los tramitados por vía prioritaria. AveXis, la división biotecnológica de Novartis AG, presentó la solicitud de autorización (Biologic License Application) en diciembre de 2018.
Peter W. Marks, director del Center for Biologics Evaluations and Research, de la FDA, declaró que, de haberse sabido la manipulación de información por parte de Novartis, la autorización probablemente se habría retrasado. Se está estudiando si esta ocultación y/o manipulación de información es susceptible de responsabilidades administrativas o penales.
El pasado martes, 6 de agosto, las acciones de Novartis AG., en Walll Street sufrieron una significativa caída. [Novartis también fabrica otro importante medicamento, Gilenya® (Fingolimod) para el tratamiento de la esclerosis múltiple].
{kind=link}
En un comunicado, Novartis dijo que respaldaba su producto. «En ningún momento durante la investigación los resultados indicaron problemas con la seguridad, eficacia o calidad del producto», dijo un portavoz de la compañía farmacéutica. «Seguimos siendo totalmente capaces de fabricar Zolgensma de alta calidad y adecuado a los niños que lo precisan».
Conceptualmente la terapia génica consiste en modificar genéticamente un virus (en este caso, un adenovirus) usándolos como transportadores de genes que se insertan y expresan en los genomas de las células que, bien carecen del gen, o portan un gen defectuoso.
La primera terapia génica autorizada por la Food and Drug Administration (FDA) estadounidense fue Luxtuma®; Zolgensma® ha sido la segunda. La biotecnología en que se fundamenta la terapia génica ha desbrozado el camino a vanguardistas tratamientos para enfermedades raras, previéndose la comercialización de entre 10 y 20 nuevas terapias cada año.
[Luxturna® (Voratigene neparvovec-rzyl) es una versión corregida del gen RPE65. Este gen codifica la síntesis de una proteína retiniana fundamental para la isomerización del retinal desde la conformación trans a la conformación cis, modificación química que forma parte de una ruta de transducción de señales que hace posible la visión. Se usa para la distrofia retiniana asociada con la mutación (no necesariamente idéntica) en las dos copias (alelos) del gen RPE65, que codifica la proteína de idéntica designación, acrónimo de Retinal Pigment Epithelium de peso molecular 65 kDalton]. [1 Dalton equivale 1 unidad de masa atómica].
El primer progreso farmacológico para la atrofia muscular espinal fue Spinraza® (Nusinersen), autorizado en el año 2016, medicamento que ha de administrarse de por vida. Por el contrario, Zolgensma®, administrado en una única perfusión, detiene la progresión de la enfermedad. Ha sido autorizado para el tratamiento resolutivo de la atrofia muscular espinal en niños menores de 2 años. El problema en estos momentos es conseguir que las compañías aseguradoras lo incluyan en su cartera de servicios, y en los criterios de selección de los niños a tratar.
La Food and Drug Administration (FDA) estadounidense ha comunicado que tuvo la primera noticia de la manipulación de datos el 28 de junio (2019), un mes después de haber aprobado Zolgensma®, a pesar de que los funcionarios de Avexis, la división de Novartis AG que desarrolló esta terapia génica conocían el problema ya en el mes de marzo.
¿Cuál fue la información manipulada?
Los problemas tuvieron que ver con experimentos en ratones durante las primeras etapas de la investigación. Una inspección realizada por la FDA entre los días 24 de julio y 2 de agosto (2019) encontró discrepancias en el mantenimiento de los registros por parte del laboratorio, así como procedimientos inadecuados en el control de calidad en la recopilación de datos experimentales. Las discrepancias más llamativas tenían que ver con la supervivencia de los animales. No hay evidencia acerca de la intencionalidad de la manipulación.
 La atrofia muscular espinal afecta a la sinapsis (placa motora) que conecta las neuronas motoras con los músculos que inervan. En el año 1992 se localizó el gen cuya mutación causa la atrofia muscular espinal. El gen se halla en el brazo corto (q) del cromosoma 5 (5q). Tres años después un grupo de trabajo dirigido por Judith Melki identificó la proteína codificada por dicho gen. La proteína se designa SMN, acrónimo de Survival Motor Neuron. Como se infiere de su denominación, esta proteína es fundamental para la supervivencia de la placa motora (sinapsis que conecta la neurona motora con el músculo esquelético inervado).
La atrofia muscular espinal afecta a la sinapsis (placa motora) que conecta las neuronas motoras con los músculos que inervan. En el año 1992 se localizó el gen cuya mutación causa la atrofia muscular espinal. El gen se halla en el brazo corto (q) del cromosoma 5 (5q). Tres años después un grupo de trabajo dirigido por Judith Melki identificó la proteína codificada por dicho gen. La proteína se designa SMN, acrónimo de Survival Motor Neuron. Como se infiere de su denominación, esta proteína es fundamental para la supervivencia de la placa motora (sinapsis que conecta la neurona motora con el músculo esquelético inervado).
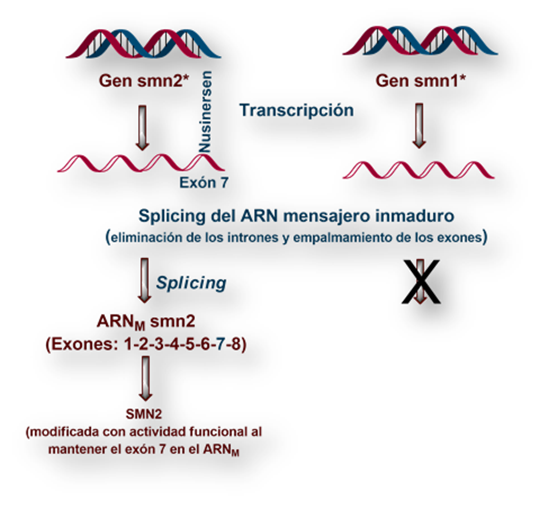
Cada una de las dos copias (alelos) del gen smn contiene información codificada para la síntesis de una de las dos versiones de la proteína SMN, esto es: SMN_1 y SMN_2. Designamos a los dos alelos como smn_1 y smn_2 respectivamente. [Los genes los escribimos con minúscula, mientras las proteínas cuya síntesis codifican los escribimos con mayúscula].
El alelo smn_1 codifica la proteína SMN_1, plenamente funcional. En cambio, el alelo smn_2 se transcribe en un ARNM que, una vez procesado (splicing), carece del exón 7. La traducción de este ARNM da lugar a una versión incompleta de la proteína SMN, a la que designamos SMN_2. Parece que el ARNM transcrito por el alelo smn_2 modula la expresión del gen que codifica la síntesis del gen smn_1.
Cuando se produce una mutación o deleción del gen smn_1 la otra copia del gen (alelo smn_2) puede suplir parcialmente esta deficiencia. De hecho, el grado de afectación de los pacientes viene determinado por la cantidad de proteína SMN_2 que se sintetiza (a mayor cantidad de proteína SMN_2, menor gravedad de la sintomatología).
La atrofia muscular espinal se manifiesta en una vez cada 11.000 nacimientos vivos, aproximadamente.
La atrofia muscular espinal se clasifica en cinco tipos:
La atrofia muscular espinal tipo 1, denominada enfermedad de Werdnig-Hoffmann, debuta alrededor de los 6 meses de edad. Cursa con hipotonía, fasciculaciones, y dificultad para deglutir y respirar. Los niños afectados no llegan a gatear, sentarse o permanecer erguidos. Tristemente suelen fallecer antes de su segundo aniversario.
La atrofia muscular espinal tipo 2 se manifiesta entre los 6 y 18 meses de edad. Los niños consiguen sentarse por sí mismos, pero son incapaces de mantenerse erguidos sin ayuda de terceros. Las infecciones son comunes. Su esperanza de vida no suele sobrepasar la adolescencia.
La atrofia muscular espinal tipo 3, llamada también enfermedad de Kugelberg-Welander, debuta entre los 2 y los 17 años. Los niños tienen una marcha anormal, dificultad para correr, levantarse de una silla, y temblor fino de los dedos. Además, padecen escoliosis (a semejanza de los niños con el «tipo 1»), y contracturas debidas a acortamiento de músculos o tendones peri-articulares. La frecuencia de infecciones respiratorias es elevada en relación a su grupo de edad y sexo. Su morbilidad es superior a la media, pero su mortalidad similar a la de la población general.
La atrofia muscular espinal con artrogriposis es una versión muy infrecuente. Las manifestaciones clínicas incluyen contracturas graves, escoliosis, deformidad del tórax, mandíbulas muy poco desarrolladas y ptosis parpebral.
La atrofia muscular espino-bulbar progresiva o enfermedad de Kennedy, puede aparecer en algún momento entre aproximadamente 15 años y la sexta década de la vida. Se manifiesta clínicamente con debilidad de los músculos de la cara, mandíbula y lengua, que trasunta en problemas de masticación, deglución y dicción. La progresión de la atrofia da lugar a fasciculaciones y alteraciones sensoriales en manos y pies debido a neuropatía sensorial (degeneración nerviosa sensorial).
La consecuencia clínica de la atrofia muscular espinal en su versión más grave («tipo 1») es la pérdida de fuerza física hasta imposibilitar la deambulación, deglución; así como la capacidad de articular sonidos y finalmente respirar de manera autónoma.
Antes de la comercialización de Spinraza® (Nusinersen) los niños con el «tipo 1» de la enfermedad fallecían antes de cumplir 2 años.
Es un asunto de enorme trascendencia cómo los sistemas de salud, públicos y privados, se adecuarán a estos innovadores y costosos tratamientos.
Zaragoza, a 9 de agosto de 2019
Dr. José Manuel López Tricas
Farmacéutico especialista Farmacia Hospitalaria
Farmacia Las Fuentes
Zaragoza