
 El neurólogo colombiano Francisco Lopera asiste a un paciente con demencia de Alzheimer
El neurólogo colombiano Francisco Lopera asiste a un paciente con demencia de Alzheimer
Los investigadores han hallado a una mujer portadora de una rara mutación genética que parece prevenir la aparición de demencia de alzhéimer.
Antecedentes-
La mujer es portadora de una mutación genética que, en principio, la predisponía a desarrollar demencia de alzhéimer a una edad relativamente joven, entre la cuarta y quinta década de vida. Pertenece a una extensa familia de la ciudad colombiana de Medellín, todos ellos portadores de una mutación genética asociada con la aparición de demencia de alzhéimer mucho antes de la senectud. La amplia familia medellinense (más de 6.000 personas) se ha convertido en un verdadero «laboratorio viviente», tras su hallazgo por el neurólogo colombiano Francisco Lopera.
La mayoría de los miembros de esta familia comienzan a sufrir alrededor de la cuarta década de vida un progresivo deterioro de la memoria y el pensamiento lógico, síntomas premonitorios de un rápido declive cognoscitivo antesala de una demencia de alzhéimer al cabo de pocos años.
Sorpresivamente, esta mujer no desarrolló los síntomas de esta grave e irreversible enfermedad neurodegenerativa; solo ahora, en su séptima década de vida, ha debutado la sintomatología.
¿Explicación?
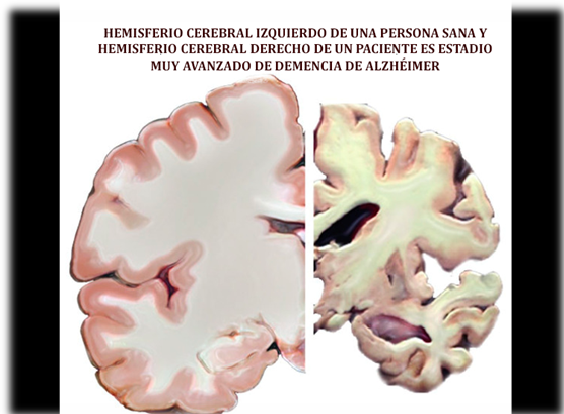
Su caso ha sido objeto de una publicación en la revista Nature Medicine. La mujer, con una mutación que le predispone a desarrollar la demencia a una edad relativamente joven, como el resto de su extensa familia, es portadora de una mutación adicional en otro gen que le protege contra las consecuencias de la primera mutación La enfermedad no se manifiesta a pesar de que su cerebro ha desarrollado un patrón neurológico concordante con la demencia de alzhéimer (profusión de placas de proteína amiloidea).
El bloqueo de la expresión del gen predisponente parece detener la progresión de la demencia. Este inesperado y sorprendente hallazgo ha abierto una nueva línea de investigación en el marasmo inextricable en que se ha convertido la farmacología de la enfermedad de alzhéimer.
Cualquier molécula que aspire a ser catalogado de medicamento ha de ensayarse previamente in vitro (cultivos de células humanas) y, posteriormente, en animales de laboratorio.
La investigación de posibles tratamientos para revertir o frenar la progresión de la demencia de alzhéimer está experimentando continuos fracasos a pesar de la inversión de inmensas sumas dinerarias. Más de 200 ensayos clínicos con potenciales fármacos han fracasado; y el último medicamento, de eficacia y duración limitada, se comercializó hace alrededor de 15 años.
Tal vez un atisbo de esperanza ha surgido con un fármaco experimental. Su designación preclínica es BAN2401 (Elenbecestat); y su efecto solo es perceptible cuando se administra a las dosis máximas toleradas. Cuando se redacta este artículo (diciembre 2019) han finalizado los ensayos clínicos fase 2, e iniciado los estudios clínicos fase 3, último requisito para su autorización por los Organismos Reguladores. La Agencia Europea del Medicamento (EMA de sus siglas en inglés, European Medicine Agency), han exigido que los pacientes con predisposición genética conocida a desarrollar la enfermedad no se incluyan en el estudio al objeto de evitar un sesgo desfavorable que podría rebajar las expectativas del estudio clínico fase 3.
Las razones por las que la investigación farmacológica sobre la enfermedad de alzhéimer está plagada de ensayos clínicos fallidos trascienden los hallazgos moleculares sobre las proteínas amiloideas y la proteína tau (ꞇ).
De una parte, no se han conseguido modelos experimentales de la enfermedad en animales de experimentación, al objeto de poder ensayar los potenciales nuevos fármacos, antes de su ensayo en humanos.
Por otra parte, los síntomas de la enfermedad debutan de manera insidiosa tras décadas de daño cerebral progresivo y silente.
Es posible, pues, que la ausencia de resultados de los medicamentos estudiados y la falta de confirmación académica de las teorías en que se sustentan se deba a que su utilización se demore demasiado, cuando los cambios cerebrales son demasiado extensos para revertir el proceso neurodegenerativo.
Por las razones expuestas previamente, algunos investigadores plantean ensayar los potenciales medicamentos ante los primeros indicios de la enfermedad; e incluso en la fase asintomática cuando las personas tienen antecedentes familiares o bien un riesgo conocido (mutaciones genéticas predisponentes). Tal vez, un criterio podría ser la determinación de los niveles de proteína amiloidea en el líquido cefálico y raquídeo. En las personas pertenecientes a grupos de alto riesgo se debería plantear si es adecuado el tratamiento profiláctico a partir de una determinada edad.
El escenario óptimo sería descubrir un marcador que se relacionase con los estadios iniciales de la enfermedad, mucho antes de que el daño cerebral sea demasiado extenso, mucho antes de los primeros signos y síntomas.
Un proyecto plantea la detección de signos tempranos de la enfermedad mediante técnicas de imaginería cerebral. En este programa, designado ADNI, acrónimo de Alzheimer’s Disease Neuroimaging Initiative, están implicados 55 centros de investigación de Estados Unidos y Canadá. Durante un sexenio se han monitorizado a 800 personas que se han sometido a escáneres cerebrales, tomografía de emisión de positrones y análisis de líquido cefálico y raquídeo. Entre los participantes había personas con lo que se denomina «deterioro cognoscitivo leve temprano», un estadio inicial de la demencia de alzhéimer. Además de mayores concentraciones de proteína amiloidea en el líquido cefálico – raquídeo, se ha observado también alteraciones del hipocampo, una región de sustancia gris diferenciada del lóbulo temporal. [El hipocampo forma parte del sistema límbico].
Volviendo a la mujer medellinense portadora de la inusual mutación, en la actualidad (diciembre 2019) tiene más de 70 años de edad. Como se ha explicado es miembro de una extensa familia (aproximadamente 6.000 personas), la mayoría afectados en edades relativamente jóvenes (4ª y 5ª décadas de vida) de demencia de alzhéimer, a la que ellos denominaban «La Bobera», que atribuyen a embrujos.
Hace algunas décadas un neurólogo colombiano, Francisco Lopera, comenzó a recopilar de manera meticulosa los registros de nacimiento y defunción de los miembros de esta familia, tanto en Medellín como en remotas aldeas. Realizó un extenso árbol genealógico, trabajando en una región donde operan guerrillas y narcotraficantes. En este entorno hostil trataba de convencer a los finados para que le entregasen los cerebros de familiares fallecidos para su estudio científico.
 Tras el análisis detallado de alrededor de 300 cerebros en la universidad colombiana de Antioquia descubrió que la elevada prevalencia de enfermedad de alzhéimer en este grupo social se debía a una mutación de un gen (PSEN1-E280A) que codifica una proteína denominada presenilina-1
Tras el análisis detallado de alrededor de 300 cerebros en la universidad colombiana de Antioquia descubrió que la elevada prevalencia de enfermedad de alzhéimer en este grupo social se debía a una mutación de un gen (PSEN1-E280A) que codifica una proteína denominada presenilina-1
La presenilina-1 es una subunidad de la proteína oligomérica secretasa-ꝩ que cataliza el clivaje de diversas proteínas en péptidos. De hecho, la presenilina-1 es la subunidad proteolítica del complejo secretasa-ꝩ, localizada en la membrana celular, donde inicia una ruta de señalización celular que lleva a cabo la transducción de señales desde la membrana al núcleo celular.
La secretasa-ꝩ ejerce una importante función en el procesamiento (proteólisis) del precursor de la proteína amiloidea. La proteína amiloidea es trascendental en la neurogénesis tanto en la etapa fetal como tras el nacimiento. Sin embargo su acumulación también se relaciona con el proceso neurodegenerativo de la enfermedad de alzhéimer.
El gen PSEN1E280A se localiza en el brazo largo (q) del cromosoma 14 en posición 24.2 (de modo abreviado: 14q24.2).
La enfermedad de alzhéimer que aflige a esta extensa familia colombiana debuta en edades tempranas al subyacer una causa genética. Este grupo poblacional es un verdadero «laboratorio viviente» donde indagar posibles remedios para esta enfermedad que afecta aproximadamente a 44 millones de personas, una cifra que se triplicará (¿?) hacia 2050, si antes no se consigue un tratamiento efectivo.
Una primera cuestión intrigante era porqué la mujer con la mutación PSEN1E280A no desarrollaba el deterioro cognitivo leve pródromo de la demencia de alzhéimer. De alguna manera, esta mujer parece ser resistente a sufrir la demencia a pesar de su especial predisposición genética.
La mujer viajó a Boston, Massachusetts, Estados Unidos, para un estudio más detallado (escáneres cerebrales y otras pruebas). En palabras de Yakeel Quiroz, neuropsicólogo colombiano, director a la sazón del laboratorio de neuroimagen en el Massachusetts General Hospital, los escáneres mostraban que su tejido cerebral contenía numerosas placas de proteína β-amiloidea, sello patognomónico de la demencia de alzhéimer. De hecho su tejido cerebral contenía muchas más placas amiloideas que otros familiares (también portadores de la mutación), probablemente porque habían fallecido antes.
A diferencia de los demás parientes, ella apenas tenía exceso de proteína tau (ꞇ), que forma anillos neurofibrilares en su cerebro. Tal vez por esta razón no tenía signos neurodegenerativos ni atrofia cerebral. Además, su escudo frente al deterioro cognitivo no ha de buscarse en su actividad intelectual (tal como nosotros la entendemos) ya que apenas tenía educación formal. Se dedicó a sacar adelante a sus cuatro hijos.
Tras la secuenciación de su genoma se descubrió que la mujer es portadora de una mutación extraordinariamente rara en un gen denominado APOE que codifica una proteína ApoE.
No está claro si la resistencia frente a la enfermedad de alzhéimer que mostraba esta mujer se debe a un retraso en el inicio del deterioro cognoscitivo leve o una especie de «congelación» de ese estadio inicial sin progresión ulterior.
 La paciente, con la mutación PSEN1E280A, predisponente a desarrollar la enfermedad de alzhéimer a edades tempranas, es también portadora homocigótica de otra rara mutación APOEch (ch, de Christchurch, patronímico de la ciudad neozelandesa donde se descubrió por primera vez). La mutación Christchurch corresponde al codón (triplete de nucleótidos) 153 que codifica el aminoácido 136, en el que la arginina ha sido sustituido por serina. Esta mutación también se presenta en otros 117 parientes de la familia medellinense, si bien todos ellos son heterocigóticos para esta mutación. Por alguna razón, el freno para la progresión a demencia de alzhéimer solo se observa cuando la mutación Christchurch se presenta en las dos copias (alelos) del gen, esto es, en homocigóticos.
La paciente, con la mutación PSEN1E280A, predisponente a desarrollar la enfermedad de alzhéimer a edades tempranas, es también portadora homocigótica de otra rara mutación APOEch (ch, de Christchurch, patronímico de la ciudad neozelandesa donde se descubrió por primera vez). La mutación Christchurch corresponde al codón (triplete de nucleótidos) 153 que codifica el aminoácido 136, en el que la arginina ha sido sustituido por serina. Esta mutación también se presenta en otros 117 parientes de la familia medellinense, si bien todos ellos son heterocigóticos para esta mutación. Por alguna razón, el freno para la progresión a demencia de alzhéimer solo se observa cuando la mutación Christchurch se presenta en las dos copias (alelos) del gen, esto es, en homocigóticos.
APO es un gen que incrementa la probabilidad de enfermedad de alzhéimer en edades tardías. Este gen (APO) tiene tres alelos: APOOE2, APOE3 y APOE4. Mientras APOE3 no influye en la predisposición a la enfermedad de alzhéimer, APOE2 disminuye el riesgo de sufrir la enfermedad a edad avanzada; y APOE4 aumenta la probabilidad en personas relativamente jóvenes. De hecho, alrededor del 40% de los enfermos de alzhéimer son portadores de la variante genética APOE4.
La mutación Christchurch modifica la «neutralidad» del gen APOE3 cuando los dos alelos están mutados. La mujer medellinense tiene dos copias mutadas del gen APOE3ch, circunstancia que parece constituir un freno a su predisposición a la demencia de alzhéimer.
Se han sintetizado moléculas que in vitro remedan la acción de esta mutación. Sería el primer paso para el desarrollo de potenciales medicamentos.
Todos los fármacos dirigidos contra las proteínas β-amiloidea y la proteína tau (ꞇ) han mostrado pobres resultados; algunos otros ni tan siquiera han superado el estadio de ensayo clínico. En estas circunstancias, una terapia genética dirigida contra el gen APOE representaría un cambio de paradigma.
Los resultados preliminares señalan que la resistencia de los homocigóticos del gen APOEch se debe a la ausencia de proteína tau (ꞇ) incluso en condiciones de elevada «carga cerebral» de proteína amiloidea (Aβ1®42).
…UN INCOVENIENTE…
Los portadores de APOEch, y otras raras mutaciones, suelen sufrir hiperlipoproteinemia tipo 3, de modo similar al 5 y 10% de los homocigóticos portadores de APOE2. Parece ser que este tipo de hiperlipoproteinemia reduce el riesgo de desarrollar demencia de alzhéimer.
Así pues, la mutación APOEch, sin ser determinista, parece ejercer una resistencia fenotípica frente al desarrollo de la enfermedad de alzhéimer.
Por otra parte, las observaciones en esta mujer (elevada presencia cerebral de proteína β-amiloidea pero sin deterioro cognoscitivo temprano) pueden llegar a poner en entredicho una creencia firmemente asentada: la vinculación entre el desarrollo de placas amiloideas en el tejido cerebral y la manifestación clínica de demencia tipo alzhéimer.
La mujer, ya en la séptima década de vida, está comenzando a manifestar un deterioro cognoscitivo importante. Antes de que no le sea posible, ha revelado el perfil genético a sus cuatro hijos, ya adultos. Todos ellos son heterocigotos para la mutación Christchurch (APOEch). Por lo tanto, no tienen la condición de homocigóticos, y, en consecuencia, carecen de la protección de su madre frente al desarrollo de la demencia.
La serendipia ha enseñado que una mujer que debería haber desarrollado demencia de alzhéimer hace más de tres décadas, ha resistido a la enfermedad durante más de treinta años, gracias a una mutación genética que «contrarresta» otra mutación predisponente. Es un descubrimiento trascendente en la frustrante investigación de medicamentos contra esta terrible enfermedad.
Zaragoza, a 19 de diciembre de 2019
Dr. José Manuel López Tricas
Farmacéutico especialista Farmacia Hospitalaria
Farmacia Las Fuentes
Florentino Ballesteros, 11-13
50002 Zaragoza