
La heparina es una sustancia compleja aislada a partir de los pulmones e intestino de cerdos y vacas.
Su actividad farmacológica consiste en retardar, incluso impedir, la coagulación sanguínea.
La interesante y compleja historia de su descubrimiento se remonta al año 1911, cuando Doyton en Francia descubrió que el extracto acuoso de hígado de perro desgrasado mostraba actividad anticoagulante. Doyton denominó a este extracto acuoso «antitrombina». Había aislado el «anticoagulante natural» que, como más tarde se descubriría, representa la diana farmacológica sobre la que actúa la heparina. Durante los siguientes tres lustros estudió su extracto sin lograr resultados significativos.
Con independencia de estos hallazgos, la verdadera historia de la heparina se inicia en el año 1916 cuando el norteamericano Mac Lean llevó a cabo investigaciones de posibles sustancias que acelerasen la coagulación.
En el año 1922, otro norteamericano, William Howell, a la sazón profesor de fisiología en la universidad John Hopkins, en Baltimore, preparó extractos similares a los que Doyton había elaborado en Francia a comienzos de siglo. Howell denominó a sus extractos heparina, término que perduró.
William Howell estudió sus extractos durante la década de 1920. Descubrió algunos aspectos de su composición química: se trataba de polisacáridos azufrados. Vendió sus primeros extractos, de un grado de pureza muy bajo, apenas el 1% o 2%, a un empresa farmacéutica de Baltimore (Hynson, Westcott and Dunning). Todos los intentos de usarlos durante trasfusiones sanguíneas fracasaron debido a la baja calidad de las preparaciones. Sin embargo, estas investigaciones atrajeron la atención de investigadores en otras partes del mundo, sobre todo Canadá y Suecia.
En el año 1928 se publicaron los primeros resultados con los extractos de Howell (Bull. John Hopkins Hosp., 1928; 42: 199-206). Durante los años siguientes, dos investigadores canadienses, David Scott y Arthur Charles, a la sazón en los Connaught Laboratories, trabajaron con objeto de lograr preparados de suficiente pureza para su empleo clínico. Finalmente, en 1933, concluyeron que la mejor fuente para la obtención del «extracto anticoagulante era el tejido pulmonar vacuno, en lugar del hígado a que referencia el epíteto heparina (J. Biol. Chem., 1933; 102: 437-48).
En 1935, Scott y Charles obtuvieron extractos de suficiente pureza para establecer el primer estándar internacional de heparina sódica.
Otros preparados obtenidos por grupos de investigación europeos (Albert Fischer, en la universidad de Copenhague; Erik Jorpes, Instituto Karolinska de Estocolmo) se ajustaban al estándar establecido en 1935.
Los primeros ensayos clínicos con heparina se realizaron en 1937, tanto en Toronto, Canadá, por Gordon Murray (prevención de trombosis tras injurias graves), como en Estocolmo, Suecia, por Clarenc Crafoord (prevención de trombosis postoperatoria). [Referencias bibliográficas: Surgery 1937; 2: 163-87 – estudio canadiense- Acta Chir Scand 1937; 79: 407-26 – estudio sueco-).
Charles Best (famoso por el descubrimiento de la insulina) comenzó a estudiar el uso de heparina para prevenir la coagulación durante las trasfusiones de sangre, técnica que sería tan útil pocos años después durante la Segunda Guerra Mundial. Así mismo, la heparina se usó para posibilitar la circulación extracorpórea; y con ella, la diálisis (1944), y el desarrollo años más tarde de la técnica de by-pass (circulación extracorpórea).
No obstante, la principal utilidad de la heparina es la prevención del tromboembolismo venoso, una de las principales causas de morbilidad y mortalidad en el mundo desarrollado.
Todavía a finales de la década de 1960, la necesidad de administrar la heparina por vía venosa, bien en Bolus frecuentes (cada 4 horas) o por perfusión continua, condicionaba su empleo.
Un importante avance se produjo a partir de 1967 cuando un equipo de investigación adscrito a los laboratorios Choay, en Francia, desarrolló una preparación de heparina en forma más concentrada que posibilitaba una administración menos frecuente, además de poder administrarse por vía subcutánea en lugar de tener que usar de modo imperativo la ruta venosa.
Así mismo, a partir de 1972. V.V. Kakkar, del King’s College Hospital de Londres demostró que bastaban pequeñas dosis de heparina para disminuir la incidencia de trombosis postoperatoria. Esta práctica se generalizó, pero con la necesidad de 2 o 3 inyecciones diarias, y riesgo de hemorragia, incluso si la dosis se ajustaba de modo adecuado.
La coagulación es un mecanismo fisiológico en el que una débil señal inicial desencadena la síntesis de cantidades muy importantes del enzima coagulante trombina. La señal que activa toda la cascada enzimática de coagulación puede ser una lesión interna (activación de la denominada «vía intrínseca») o una señal externa («vía extrínseca», generalmente el contacto con una superficie con carga negativa neta, tal como el vidrio de un tubo de ensayo). En cualquier caso (vías intrínseca o extrínseca) dan lugar a la transducción de una débil señal inicial en una masiva formación de la enzima trombogénica trombina.
¿Cómo consigue la heparina retardar la coagulación sanguínea?



La cascada de coagulación tiene sus propios mecanismos de retroalimentación. El moderador de la coagulación es la antitrombina (AT-III). Esta proteína inhibe lentamente la mayor parte de los factores enzimáticos de la coagulación, en especial la trombina (factor IIa) y la fibrina (factor Xa). R.D.Rosenberg, demostró en 1973 que la heparina se une a la AT-III. El complejo [heparina↔antitrombina] así formado inhibe los factores de coagulación unas 103 veces más rápido que la AT-III libre (no unida a la molécula de heparina).
Así pues, la heparina aumenta la actividad del principal inhibidor de la coagulación, la antitrombina (AT-III). La heparina actúa pues como un activador del inhibidor.
Hasta la década de 1970 las actividades antitrombótica y anticoagulante de la heparina se consideraban indisociables.
La situación cambió a partir de 1976, con el perfeccionamiento de la tecnología de separación de mezclas de macromoléculas en función de su peso molecular.
E.A. Johnson en Reino Unido llevó a cabo los primeros fraccionamientos de la molécula de heparina. Demostró de esta manera que los fragmentos «de bajo peso molecular» tenían una fuerte actividad anti-factor-Xa (factor Stuart-Prower), pero carecían de efecto sobre el factor IIa (trombina).
Por el contrario, la heparina «de elevado peso molecular» (no-fraccionada) conserva las dos actividades (anti-factor-Xa y anti-factor-IIa).
La inhibición del factor IIa (trombina) es responsable de la acción anticoagulante de la heparina, mientras que la acción antitrombótica depende también de la actividad anti-factor-Xa. De esta manera, seleccionando el fragmento de heparina en base a su peso molecular se podían obtener preparados con actividad específica anticoagulante o inespecífica (anticoagulante y antitrombótica).
Un equipo de investigación de laboratorios Choay (hoy integrados en la multinacional francesa Sanofi Aventis) logró en 1978 un «nuevo» medicamento preparado a partir de fragmentos de heparina «de bajo peso molecular». Se consiguió una preparación de heparina con elevada actividad anticoagulante y una actividad antitrombótica residual. La relación de actividades anticoagulante vs antitrombótica era de: 4:1.
La heparinas «de bajo peso molecular» (que han terminado por desplazar a las heparinas no-fraccionadas de la praxis médica) se comercializaron a partir de la segunda mitad de la década de 1980 (Eur. Pat. 1981; 40144). Estas heparinas «de bajo peso molecular» consiguen suficiente protección frente a la enfermedad tromboembólica reduciendo el riesgo de hemorragia asociado al efecto anticoagulante. Otras importantes ventajas son una mejor biodisponibilidad siguiendo su inyección subcutánea.
Una inyección subcutánea diaria de heparina «de bajo peso molecular» lograba una protección frente al riesgo de trombosis equiparable al logrado con la administración de dos o tres inyecciones diarias de las heparinas no-fraccionadas.
Choay (entonces perteneciente a Rhône-Poulenc) lanzó al mercado farmacéutico mundial la primera heparina «de bajo peso molecular» en el año 1986. Le siguieron pronto otros laboratorios, con sus propios preparados comerciales.
La investigación ulterior se dirigió a la obtención de fragmentos más pequeños de la heparina, por lo tanto más específicos en su unión a la antitrombina, a la vez que se investigaba en qué fragmento de la molécula de heparina residía su acción farmacológica (unión a la antitrombina).
Tres grupos de investigación, trabajando independientemente (R.D. Rosenberg en Estados Unidos y U. Linhald y L.O. Anderson, en Suecia) observaron que solo una tercera parte de las moléculas de heparina tenían la facultad de asociarse a la antitrombina. Las investigaciones, en las que también participó el Instituto Choay de Francia y el grupo italiano de B. Casu, identificaron la estructura química de la molécula de heparina que se engarza con la antitrombina: se trata de un pentasacárido.
El pentasacárido se sintetizó finalmente en los laboratorios Choay en 1983. Los ensayos realizados confirmaron que, efectivamente, este pentasacárido era la región de la molécula de la heparina que se enlaza con la antitrombina, activándola.
Enseguida se emprendió, dentro del programa Eureka, que asociaba el grupo Sanofi (hoy día Sanofi Aventis, al que también pertenece el Instituto Choay) y el grupo holandés Akzo de los laboratorios Organon, la síntesis de laboratorio de este pentasacárido.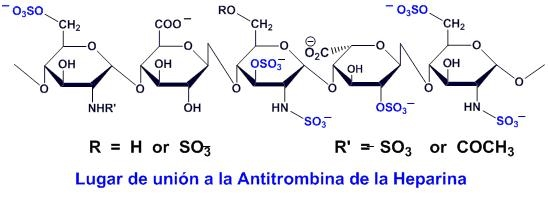
Todavía más: el grupo de R.D. Rosenberg descubrió en 1985 que este pentasacárido tapiza el endotelio vascular evitando así la trombosis espontánea en condiciones fisiológicas.
Resumen cronológico de los hitos más importantes en el descubrimiento de la heparina
- 1916: Mac Lean (primeras investigaciones estandarizadas).
- 1922: William Howell (primer uso del término heparina).
- 1928: David Scott y Arthur Charles (Connaugth Laboratories, Canadá) (purificación de los extractos).
- 1935: 1er estándar internacional de heparina sódica (David Scott y Arthur Charles, Canadá; Albert Fischer (Copenhague, Dinamarca); Erik Jorpes (Karolinska Instituten, Estocolmo, Suecia).
- 1937: -Gordon Murray (Canadá) y Clarenc Crafoord (Suecia): primeros ensayos en humanos.
- 1944: Inicio de la diálisis (la heparina era fundamental para su práctica).
- 1972: V. V. Kakkar (King’s College, Londres): primeras heparinas «de bajo peso molecular» (experimentales).
- 1973: R.D. Rosenberg (USA) (mecanismo de acción de la heparina. Disociación de las actividades antitrombótica y anticoagulante).
- 1978: Grupo Choay (Francia): desarrollo de la primera heparina «de bajo peso molecular».
- 1983: Síntesis del pentasacárido (sitio activo de la heparina en su unión a la antitrombina).
- 1986: Rhône-Poulenc (hoy integrado en Sanofi Aventis) comercializó la primera heparina de bajo peso molecular (heparina fraccionada).
Las funciones de la heparina van mucho más allá de su ya de por sí trascendente actividad anticoagulante (heparinas fraccionadas) y antitrombótica y anticoagulante (heparina no-fraccionada).
La heparina provoca la liberación de la enzima superóxido-dismutasa para la eliminación de los radicales libres; desencadena la secreción del «factor activador del plasminógeno» (inductor de la disolución de los coágulos sanguíneos); inhibe la secreción de la enzima heparanasa, involucrada en la aparición de metástasis, tal vez mediante la regulación del crecimiento y diferenciación celular durante la angiogénesis.
La heparina no es una sustancia farmacológicamente agostada. Diversos grupo de investigación, en particular los de M.J. Karnovsky y G.L. Nicolson en Estados Unidos, I. Vlodavsky en Israel, y E. de Clercq en Bélgica, continúan buscando nuevas aplicaciones de esta maravillosa sustancia.
Zaragoza, a 29 de marzo de 2019
Dr. José Manuel López Tricas
Farmacéutico especialista Farmacia Hospitalaria
Farmacia Las Fuentes
Florentino Ballesteros, 11-13
50002 Zaragoza