
La enfermedad de Pompe es una de las patologías hereditarias que afectan al almacenamiento corporal del glucógeno, un polímero de glucosa. El glucógeno es la forma en que el organismo almacena energía metabólica de utilización inmediata. [El metabolismo de las grasas – lípidos – es mucho más lento].
Otras glucogenopatías son las enfermedades de von Gierke, Cori, Andersen, McArdle, Hers; y otras dos designadas como glucogenopatías VII y VIII.
En su versión más grave, la enfermedad de Pompe no tratada es mortal antes del primer año de vida.
Se distinguen tres tipos de enfermedad de Pompe, en función de la fase vital a la que debuta, que determina, a su vez, la prognosis. Son éstos: la «infantil clásica» (la forma más grave, con una supervivencia sin tratamiento inferior al primer año de vida); la «infantil no-clásica» o «juvenil»; y la «inicio tardío» (o «adulto»).
La forma «infantil clásica» debuta a los pocos meses del nacimiento. La fase prodrómica consiste en debilidad muscular (miopatías), falta de tono muscular (hipotonía), hepatomegalia y defectos cardíacos evidenciados radiológicamente por cardiomegalia. Los niños no ganan peso siguiendo el patrón normal de crecimiento, y comienzan a manifestar problemas respiratorios.
La versión de la enfermedad de Pompe «infantil no-clásica» debuta alrededor del primer aniversario de vida. Se caracteriza por un retraso en la adquisición de habilidades motoras (girar el cuerpo y sentarse), pero, a pesar de la cardiomegalia, la función cardíaca se mantiene. Por el contrario, los problemas respiratorios son más graves; y los niños afectados raramente sobreviven más allá de la pubertad.
La enfermedad de Pompe de «inicio tardío» se manifiesta más tardíamente, entre el final de la infancia y los primeros años de la madurez. La sintomatología es más leve que las versiones infantiles. En la forma «adulta» (de «inicio tardío»), no se observa cardiopatía. La manifestación clínica más evidente es la debilidad muscular, sobre todo de piernas y tronco, siendo la miopatía e hipotonía de los músculos respiratorios el aspecto más comprometedor. De hecho, el cuadro clínico deriva irremisiblemente a una grave insuficiencia respiratoria.
 La enfermedad de Pompe está causada por una mutación en el gen GAA, que codifica la síntesis de la enzima maltasa (técnicamente α-glucosidasa). La maltasa se localiza en los lisosomas, orgánulos sub-celulares que actúan como fábricas de reciclaje de los productos y subproductos del metabolismo. En los lisosomas el glucógeno se hidroliza en monómeros de glucosa, la principal fuente metabólica de las células.
La enfermedad de Pompe está causada por una mutación en el gen GAA, que codifica la síntesis de la enzima maltasa (técnicamente α-glucosidasa). La maltasa se localiza en los lisosomas, orgánulos sub-celulares que actúan como fábricas de reciclaje de los productos y subproductos del metabolismo. En los lisosomas el glucógeno se hidroliza en monómeros de glucosa, la principal fuente metabólica de las células.
Las mutaciones del gen GAA (localizado en el brazo largo del cromosoma 17, esto es, 17q) dan lugar a que el gen modificado sintetice una α-glucosidasa que no puede llevar a cabo su actividad enzimática de hidrólisis del glucógeno. La acumulación del polímero daña gravemente las células, siendo más evidente en las células musculares, que constituyen verdaderos almacenes de glucógeno en el organismo.
La enfermedad de Pompe, al igual que otras genopatías del metabolismo del glucógeno, se heredan siguiendo un modelo autosómico recesivo, esto es, se trata de una herencia no vinculada con cromosomas sexuales (herencia autosómica); y en la que los dos alelos (copias de un gen) deben estar mutadas para que se manifieste en toda su intensidad la clínica de la enfermedad (condición de «recesividad»).
Existe un tratamiento: α-glucosidasa humana recombinante (designada como rhGAA) denominada internacionalmente como alglucosidasa-α (Myozyme®). Sin embargo, algunos pacientes desarrollan anticuerpos contra este fármaco, el único que puede salvarles la vida. Este escenario clínico en el que los pacientes desarrollan anticuerpos contra los medicamentos (generalmente macromoléculas) que se les administra, es mucho más común de lo que a priori pudiera creerse. [Myozyme® se comercializa como viales de 20ml conteniendo 50mg de alglucosidasa-α. El vial se reconstituye para su infusión IV].
De sólito, los anticuerpos se desarrollan contra macromoléculas (o fragmentos de éstas denominados epítopos) anclados sobre la superficie de los virus o la membrana de bacterias. Pero el sistema inmune también fabrica anticuerpos contra grandes moléculas, tales como algunos medicamentos con estructura proteica.
Los anticuerpos fabricados por el sistema inmune que se dirigen contra un medicamento específico (que se comporta cual si fuese un antígeno) pueden neutralizar su efecto completamente. Sin embargo, no existe forma de prever en qué pacientes ni bajo qué circunstancias sucederá.
En un interesante trabajo publicado en marzo (2017) en The New England Journal of Medicine, la multinacional norteamericana Pfizer dio cuenta de que en los estadios finales de un ensayo clínico de un nuevo medicamento para la hipercolesterolemia (Bococizumab), muchos de los 30.000 participantes dejaron de responder a la acción del fármaco estudiado.
Al principio los participantes en el ensayo clínico respondieron muy favorablemente al tratamiento; sus niveles de colesterol se redujeron de modo drástico, pero más adelante las concentraciones plasmáticas de colesterol comenzaron a aumentar. El fármaco parecería que hubiese dejado de hacer efecto.
Pfizer se vio obligado a interrumpir el ensayo con el anticuerpo monoclonal Bococizumab, tras una inversión de billones de dólares. Otros dos anticuerpos monoclonales similares a Bococizumab, Evolocumab (de Amgen) y Arilocumab (comercializado conjuntamente por Sanofi Aventis y Regeneron Pharmaceuticals) se comercializan desde hace algún tiempo, y son prescritos sin aparentes problemas de eficacia.
Steve Danehy, portavoz de Pfizer, declaró que hasta un 87% de los pacientes que toman anticuerpos monoclonales terminarán desarrollando anticuerpos que comprometerán o anularán la eficacia clínica.
Ante este escenario, pacientes y clínicos se hallan inoperantes. Por ahora solo cabe sustituir el fármaco por otro similar (otro anticuerpo monoclonal) y confiar que funcione, pero siempre con la expectativa de que el paciente terminará, con elevada probabilidad, desarrollando anticuerpos contra este nuevo medicamento.
Sin embargo, este modo de proceder solo es factible cuando existen varias alternativas terapéuticas. No es así en las situaciones en las que solo existe una opción terapéutica. Tal es el caso de la enfermedad de Pompe.
En un pequeño pero significativo subgrupo de pacientes con gota, en los que la enfermedad es extremadamente grave, los anticuerpos que bloquean la única alternativa farmacológica, anulan cualquier opción de tratamiento.
Los niños con la genopatía de Pompe nacen aparentemente sanos. A los pocos meses de vida manifiestan lo que al principio parece un resfriado que no remite en plazo. Cuando se realiza un examen radiográfico, la primera y llamativa observación es un aumento del tamaño del corazón (cardiomegalia). Tras la confirmación del diagnóstico (enfermedad de Pompe), se instauran infusiones intravenosas quincenales de la enzima humana recombinante alglucosidasa (1,6-α-glucosidasa) (abreviadamente rhGAA), comercializada desde el año 2006.
Las primeras infusiones logran mejorías. Los niños aprenden a sentarse y a utilizar sus brazos, entre otras funciones básicas propias de su corta edad. Pero, en algunos casos, tras varias infusiones las mejorías se detienen. Sucede como si el medicamento hubiese dejado de funcionar. El problema es enorme: si se interrumpen las infusiones, la situación clínica progresará hasta una grave insuficiencia respiratoria que requerirá traqueotomía, y fallo cardíaco irreversible. Todo ello en un escenario donde los pequeños progresos del desarrollo se diluirán rápidamente.
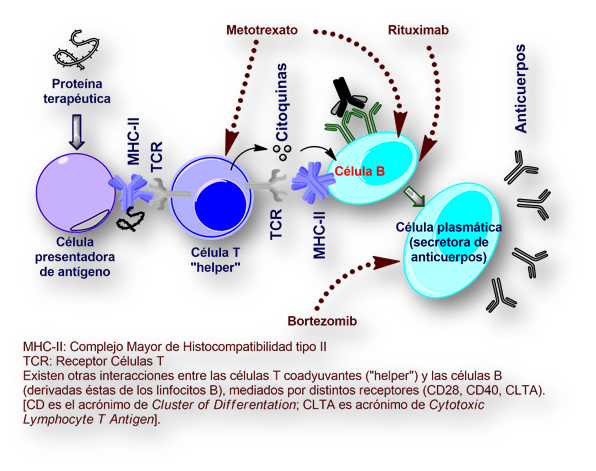 La estrategia ensayada consiste en la administración de un complejo cóctel farmacológico que incluye un antiguo medicamento, Metotrexato, junto a otro anticuerpo monoclonal (Rituximab), y Bortezomib. Junto a esta estrategia de anulación controlada del sistema inmune se infunden gammaglobulinas para no dejar indefenso al paciente contra las infecciones.
La estrategia ensayada consiste en la administración de un complejo cóctel farmacológico que incluye un antiguo medicamento, Metotrexato, junto a otro anticuerpo monoclonal (Rituximab), y Bortezomib. Junto a esta estrategia de anulación controlada del sistema inmune se infunden gammaglobulinas para no dejar indefenso al paciente contra las infecciones.
[Metotrexato es un inhibidor de la enzima «dihidrofolato-reductasa», que cataliza la adición de cuatro equivalentes de reducción necesarios para la conversión del folato, primero en dihidrofolato, y a continuación de tetrahidrofolato].
[Rituximab es un anticuerpo monoclonal quimérico dirigido contra el antígeno (receptor linfocitario) CD20].
[Bortezomib es un inhibidor reversible del proteosoma, un complejo proteico responsable de la degradación controlada de las proteínas celulares durante el ciclo celular. Su denominación común internacional hace referencia a la presencia de un átomo de boro en su estructura molecular].
Esta estrategia para el tratamiento usada en niños que han desarrollado anticuerpos contra el único medicamento disponible (la hormona deficitaria obtenida por ingeniería genética), se puede extrapolar a otras enfermedades en las que los afectados desarrollan anticuerpos contra las medicaciones que precisan.
Un ensayo clínico realizado por Selecta Biosciences está estudiando un preparado farmacéutico en el que un fármaco «supresor de anticuerpos» se empaqueta en nano-partículas biodegradables, que se administran junto con el medicamento que precisa el paciente.
Otro enfoque al problema es el que está llevando a cabo el Dr. Paul M. Ridker, del Brigham and Women’s Hospital, en Boston, Massachusetts, Estados Unidos. El trabajo, financiado por Pfizer, fabricante del Bococizumab, trata de desentrañar de qué manera se puede conocer con anticipación los pacientes que desarrollarán anticuerpos contra un determinado medicamento macromolecular. Esta estrategia sería muy útil para seleccionar correctamente a los pacientes antes de instaurar determinadas terapias; y, así mismo, retomar el uso de fármacos que, como ha sucedido con Bococizumab, han visto interrumpidos sus estudios clínicos.
Zaragoza, a 23 de mayo de 2017
Dr. José Manuel López Tricas
Farmacéutico especialista Farmacia Hospitalaria
FARMACIA LAS FUENTES
ZARAGOZA