
Imagen computerizada del cerebro de un paciente con adrenoleucodistrofia donde se evidencia la progresiva desmielinización (A ® B) (pérdida de sustancia blanca).
La adrenoleucodistrofia (ADL) es una gravísima enfermedad, por su sintomatología e irreversibilidad, causada por una mutación en el cromosoma X. Se engloba en un conjunto de enfermedades neurodegenerativas denominadas genéricamente leucodistrofias. Todas ellas comparten una característica anatómico-patológica: la desaparición de la envoltura de mielina de muchos axones del sistema nervioso central y periférico. Esta pérdida de mielina tiene su trasunto en graves e irreversibles alteraciones neurológicas.
Al tratarse de una mutación del cromosoma X, las mujeres actúan como portadoras y perpetuadoras de la genopatía. Los hijos varones (XY) de una madre portadora tienen una probabilidad del 50% de padecer la enfermedad si heredan la copia (alelo) defectuoso del cromosoma X. Más específicamente, el gen mutado se designa como ABCD1. Se localiza en el brazo corto (q) del cromosoma X, en la posición 28 a partir del centrómero. De modo abreviado: Xq28.
La prevalencia de adrenoleucodistrofia es de 1 caso por cada 21.000 nacimientos vivos.
Los enfermos de adrenoleucodistrofia (ADL) acumulan ácidos grasos de cadena muy larga, sobre todo en el cerebro y las glándulas adrenales. El gen (carencial o no-funcional) de los afectados por la enfermedad codifica una proteína que escinde los ácidos grasos de cadena muy larga, para su posterior oxidación metabólica (β-oxidación).
Una consecuencia de la acumulación de ácidos grasos de cadena muy larga en la región cortical de las glándulas adrenales es la destrucción de las células que sintetizan y segregan hormonas esteroides fundamentales para el metabolismo. Se desarrolla enfermedad de Addison.
La adrenoleucodistrofia debuta durante la infancia, entre los 4 y 14 años de edad. Los primeros síntomas se pueden confundir con problemas de adaptación escolar (retraimiento, comportamientos agresivos catalogados de rabietas infantiles). Pronto aparecen otros síntomas más graves, tales como pérdida de memoria y alteraciones del equilibrio. El cuadro patognomónico se complica con alteraciones visuales, auditivas, ataxia. fatiga, vómitos intermitentes, excesiva pigmentación de la piel y signos de demencia. La supervivencia desde el diagnóstico no suele sobrepasar un lustro.
Una enfermedad similar, pero de inicio tardío (entre los 21 y los 35 años aproximadamente), denominada adrenomieloneuropatía (AMN) cursa con rigidez, debilidad, parálisis de los miembros inferiores y ataxia. La progresión, aun cuando también se acompaña de moderado deterioro neurológico, es menos dramática. El 50% de las mujeres portadoras del gen de la adrenoleucodistrofia (ADL) desarrollarán una clínica moderada de adrenomieloneuropatía (AMN).
La «adrenoleucodistrofia neonatal» es una enfermedad estrechamente relacionada pero de aparición muy temprana (neonatos).
Hasta ahora los únicos tratamientos para la adrenoleucodistrofia eran, bien un trasplante de médula ósea de un donante compatible; o un trasplante de sangre del cordón umbilical si éste se había conservado al nacer. No obstante ambas técnicas son onerosas y bastante peligrosas, con una mortalidad de aproximadamente el 20%. Además, los que sobreviven arrastran de por vida graves discapacidades.
Sin embargo, un estudio (Starbean Study) publicado online en la revista The New England Journal of Medicine, ha dado a conocer un tratamiento mediante una terapia génica (designada como Lenti-D™), carente de efectos adversos, pero con una limitación: ha de aplicarse muy temprano, cuando aparecen los signos premonitorios de la enfermedad, solo detectables mediante escáneres cerebrales.
El estudio ha involucrado a 17 niños y adolescentes con edades comprendidas entre los 4 y 13 años de edad. Todos recibieron la terapia génica experimental. Dos años más tarde, 15 de ellos estaban normales sin síntomas objetivos de la enfermedad.
De los dos muchachos que no respondieron, uno de ellos murió porque la enfermedad progresó tan rápidamente que nada pudo hacer la terapia génica. El otro abandonó el estudio para someterse a un trasplante de médula ósea, falleciendo por las complicaciones sufridas.
El estudio abre inusitadas esperanzas para la generalización de terapias génicas en enfermedades cerebrales hasta ahora irresolubles.
La investigación que condujo al desarrollo de la terapia génica contra la adrenoleucodistrofia comenzó con Amber Salzmann, una ejecutiva con doctorado en matemáticas, que trabaja para la multinacional farmacéutica GlaxoSmithKline Pharma. En el año 2000 su sobrino recibió el diagnóstico de adrenoleucodistrofia. Hasta entonces sus únicas referencias de la enfermedad procedían de una famosa película Lorenzo’s Oil, «El aceite de la vida» en su versión española.
Cuando decidieron utilizar la terapia génica en la adrenoleucodistrofia, se enfrentaron a dos antecedentes muy desfavorables. Hacía poco que la terapia génica había caído en desgracia después de que Jesse Gelsinger, de 18 años, muriese durante un tratamiento experimental contra la deficiencia de ornitina-transcarbamilasa. Además, en el año 2003, cuatro de nueve niños que recibieron terapia génica para una inmunodeficiencia desarrollaron leucemia.
No obstante los antecedentes, se optó por una apuesta arriesgada: utilizar el VIH (Virus de Inmunodeficiencia Humana) desactivado como portador del gen ausente o defectuoso en el genoma de los niños con adrenoleucodistrofia. El VIH es, hoy por hoy, el mejor transportador de genes a las células humanas. Pero su sola mención obliga a adoptar medidas de seguridad extraordinarias.
Los primeros estudios se realizaron en Francia. Los investigadores modificaron una versión desactivada del VIH, insertando en el genoma vírico el gen ausente o defectuoso en los pacientes con adrenoleucodistrofia.
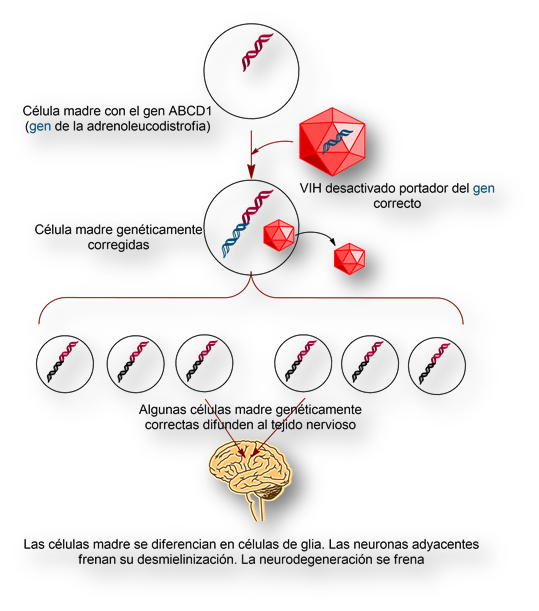 El procedimiento consiste en extraer células madre de la médula ósea de niños con adrenoleucodistrofia. A continuación se inserta el gen deficitario en el genoma de las células madre, usando el VIH desactivado como vector. Finalmente las células madre, genéticamente corregidas, se inyectan de nuevo en el niño. Estas células madre corregidas comienzan un largo proceso de multiplicación en la médula ósea. Al cabo de 1 año aproximadamente algunas células difunden en el tejido nervioso especializándose en células de glía. Estas células, genéticamente correctas, aíslan las neuronas adyacentes, bloqueando la desmielinización de sus axones; y, en última instancia, deteniendo el proceso neurodegenerativo. Es como si las células de glía genéticamente corregidas tomaran el control sobre las neuronas adyacentes, bloqueando su degeneración (desmielinización).
El procedimiento consiste en extraer células madre de la médula ósea de niños con adrenoleucodistrofia. A continuación se inserta el gen deficitario en el genoma de las células madre, usando el VIH desactivado como vector. Finalmente las células madre, genéticamente corregidas, se inyectan de nuevo en el niño. Estas células madre corregidas comienzan un largo proceso de multiplicación en la médula ósea. Al cabo de 1 año aproximadamente algunas células difunden en el tejido nervioso especializándose en células de glía. Estas células, genéticamente correctas, aíslan las neuronas adyacentes, bloqueando la desmielinización de sus axones; y, en última instancia, deteniendo el proceso neurodegenerativo. Es como si las células de glía genéticamente corregidas tomaran el control sobre las neuronas adyacentes, bloqueando su degeneración (desmielinización).
En los primeros ensayos, esta terapia génica contra la adrenoleucodistrofia ha superado todas las expectativas de los investigadores, lográndose detener la degeneración cerebral de los niños afectados.
No obstante, la terapia génica ha de realizarse muy prontamente porque el periodo de latencia hasta observar resultados (1 año aproximadamente) puede ser excesivo en niños con sintomatología muy evidente. En estos casos, la degeneración puede haber alcanzado el umbral de no-retorno.
El inesperado éxito de este pequeño estudio piloto ha sido suficiente para crear la compañía Bluebird Bio, que espera comercializar la primera terapia génica contra la adrenoleucodistrofia.
Bluebird Bio ha ampliado el estudio a otros ocho niños, comparando los resultados con un grupo de niños a los que se sometió a trasplante de médula, actuando éstos a modo de «grupo control».
El precio de la potencial nueva terapia génica se hará público cuando sea autorizada. Los expertos asumen que su coste será similar al de un trasplante de médula ósea (cientos de miles de dólares). Puede parecer muy elevado, pero considérese que la terapia génica, a diferencia de los trasplantes de médula ósea, parece ser un tratamiento curativo.
Zaragoza, a 17 de octubre de 2017
Dr. José Manuel López Tricas
Farmacéutico especialista Farmacia Hospitalaria
Farmacia Las Fuentes
Zaragoza